1.本发明涉及一种新型高效2,3,5,6-四氟苄醇的制备工艺,属于精细化工技术领域。
背景技术:
2.2,3,5,6-四氟苄醇是制备四氟苯菊酯的关键中间体。四氟苯菊酯属于广谱杀虫剂,急慢性毒性极低,广泛应用于卫生杀虫制品,是对环境友好的绿色农药。文献报道2,3,5,6-四氟苄醇的制备工艺主要有六条路线。
3.路线一:以2,3,5,6-四氟对苯二甲酸二甲酯为原料制备2,3,5,6-四氟苄醇。2,3,5,6-四氟对苯二甲酸二甲酯在乙二醇二甲醚溶剂中硼氢化钠还原,还原所得灰白色固体水解后得到2,3,5,6-四氟对羟甲基苯甲酸(收率82.6%)和2,3,5,6-四氟对二苯甲醇(收率11.57%)。2,3,5,6-四氟对羟甲基苯甲酸在dmso中脱羧得到2,3,5,6-四氟苄醇。该工艺原料2,3,5,6-四氟对苯二甲酸甲酯由2,3,5,6-四氟对苯二腈经水解和甲酯化制备。该路线若以2,3,5,6-四氟对苯二腈计实际上需经五步反应完成,路线较长,经济性不佳。代表文献有cn201711008251。
4.路线二:以2,3,5,6-四氟苯甲酸为原料制备2,3,5,6-四氟苄醇。de3714602以硼氢化钠为还原剂/硫酸二甲酯为活化剂一步还原得到2,3,5,6-四氟苄醇。cn1900037以硼氢化钠为还原剂,氯化锌为活化剂,一步还原得到四氟苄醇。以上工艺均需用到大量较为昂贵还原剂硼氢化钠,有的工艺甚至用到10当量硼氢化钠,导致生产成本较高。
5.路线三:以2,3,5,6-四氟苯甲酰氯为原料制备2,3,5,6-四氟苄醇。cn2006101010646和cn109293478等以2,3,5,6-四氟苯甲酰氯为原料乙二醇二甲醚为溶剂,硼氢化钠还原得到2,3,5,6-四氟苄醇。2,3,5,6-四氟苯甲酰氯用2,3,5,6-四氟苯甲酸与二氯亚砜反应制备,在工艺步骤上要比直接使用2,3,5,6-四氟苯甲酸为原料多一步。
6.路线四:以2,3,5,6-四氟苯甲酸甲酯为原料制备2,3,5,6-四氟苄醇。《有机合成》2005,25,1125报道以2,3,5,6-四氟苯甲酸甲酯为原料在碘单质催化下,硼氢化钠还原得到2,3,5,6-四氟苄醇。该反应条件较为温和,然而除使用大量硼氢化钠外,还用到了大量昂贵碘单质。
7.路线五:以1,2,4,5-四氟苯为原料制备2,3,5,6-四氟苄醇。cn113292399报道了将1,2,4,5-四氟苯与四氯化碳傅克反应,得到2,3,5,6-四氟三氯甲苯后,2,3,5,6-四氟三氯甲苯再催化水解得到2,3,5,6-四氟苯甲酰氯,2,3,5,6-四氟苯甲酰氯经罗森蒙德还原得到2,3,5,6-四氟苯甲醛,然后2,3,5,6-四氟苯甲醛用pt/mgal2o4为催化剂加氢还原得到2,3,5,6-四氟苄醇。该路线使用了市场难以购得四氯化碳和市场规模产量较小1,2,4,5-四氟苯,用到了昂贵的催化剂pd和pt,而且罗森蒙德还原和醛还原为醇这两步反应均用到了氢气,存在较大安全隐患。因而,该路线在成本方面和安全控制上均不具有优势。
8.路线六:以五氟苯腈为原料制备2,3,5,6-四氟苄醇。cn101462928报道五氟苯腈在活泼金属作用下脱氟得到2,3,5,6-四氟苯腈,然后经高压催化加氢得到2,3,5,6-四氟苄
胺,2,3,5,6-四氟苄胺再进行重氮化水解后得到2,3,5,6-四氟苄醇。或五氟苯腈先进行催化加氢得到五氟苄胺,五氟苄胺重氮化水解后得到五氟苄醇,五氟苄醇再用活泼金属脱氟得到2,3,5,6-四氟苄醇。该路线以五氟苯腈为原料经三步反应得到2,3,5,6-四氟苄醇,然而将氰基转化为苄胺需要在高压条件下催化加氢才可实现,氢压高达10-20atm。高压加氢存在极大安全隐患。其次将苄胺转化为苄醇是在重氮化条件下实现的,重氮化会产生大量高盐废水。此外,用活泼金属对五氟苄醇进行脱氟时大量活泼金属与酸反应产生氢气,容易发生爆炸和火灾。
技术实现要素:
9.为了解决上述技术问题,本发明的目的在于提出一种安全可控、绿色环保、成本低、高纯度2,3,5,6-四氟苄醇的制备工艺。本发明的技术方案为:
10.一种2,3,5,6-四氟苄醇的制备工艺,其特征在于,包括以下步骤:
11.第一步,五氟苯腈对位脱氟:以五氟苯腈为原料在甲酸和甲酸钠体系中,在催化剂存在下反应得到2,3,5,6-四氟苯腈;
12.第二步,stephen还原:2,3,5,6-四氟苯腈在含有氯化氢的醚类溶剂中,二氯化锡存在下还原得到2,3,5,6-四氟苯甲醛;
13.第三步,硼氢化物还原:2,3,5,6-四氟苯甲醛在含醇类溶剂中,硼氢化物还原得到2,3,5,6-四氟苄醇。
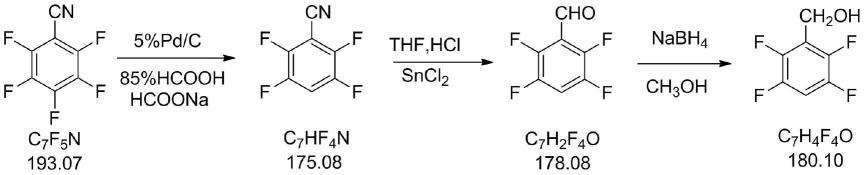
14.反应方程式表示为:
[0015][0016]
进一步地,在上述技术方案中,第一步操作为,向85%甲酸中加入五氟苯腈和二水甲酸钠后加热,然后再加入催化剂反应;反应完毕回收催化剂和甲酸;加水后含氯溶剂提取水相;有机相常压回收含氯溶剂后减压蒸馏得到2,3,5,6-四氟苯腈。
[0017]
进一步地,在上述技术方案中,第一步中,催化剂选自pd/c或pt/c,含量为0.1%~20%;催化剂与五氟苯腈质量比为0.01~1:1。
[0018]
进一步地,在上述技术方案中,第一步中,甲酸与五氟苯腈质量比为5-50:1;甲酸钠与五氟苯腈质量比为1-20:1;反应温度为50-80℃。
[0019]
进一步地,在上述技术方案中,第二步操作为,向冷却至-10℃~10℃醚类溶剂中通入干燥氯化氢气体并使其饱和;加入二氯亚锡后分批加入2,3,5,6-四氟苯腈进行还原;反应结束,含氯溶剂提取;有机相常压回收含氯溶剂再减压精馏得到2,3,5,6-四氟苯甲醛。
[0020]
进一步地,在上述技术方案中,第二步中,醚类溶剂选自甲基四氢呋喃、环戊基甲基醚、乙二醇二甲醚或叔丁基甲基醚;醚类溶剂与2,3,5,6-四氟苯腈质量比为1-50:1;反应温度为0~50℃。
[0021]
进一步地,在上述技术方案中,第三步操作为,2,3,5,6-四氟苯甲醛溶于含醇类溶剂中,控温0-50℃分批加入硼氢化物;反应完毕减压回收溶剂后加水酸化,含氯溶剂提取料
液;有机相常压回收含氯溶剂,减压精馏得到2,3,5,6-四氟苄醇。
[0022]
进一步地,在上述技术方案中,第三步中,硼氢化物为硼氢化钠、硼氢化钾或硼氢化锂;硼氢化物与2,3,5,6-四氟苯甲醛摩尔比为0.25-10:1。
[0023]
进一步地,在上述技术方案中,第三步中,醇类溶剂选自甲醇、乙醇或异丙醇,含醇类溶剂为醇类溶剂或醇类溶剂与甲苯、二氯甲烷的混合溶剂。
[0024]
进一步地,在上述技术方案中,含氯溶剂选自二氯甲烷、氯仿或1,2-二氯乙烷。
[0025]
发明有益效果
[0026]
本发明提出的一种高效2,3,5,6-四氟苄醇的制备工艺以五氟苯腈为起始物料,经钯碳对位脱氟、stephen还原、硼氢化钠将醛还原为醇得到2,3,5,6-四氟苄醇。五氟苯腈市场易得,且已形成较大的生产规模。五氟苯腈在甲酸和甲酸钠体系中采用5%pd/c进行对位脱氟得到2,3,5,6-四氟苯腈,具有较好的选择性。stephen还原是指在甲基四氢呋喃氯化氢溶液中用二氯亚锡还原将氰基转化为醛基。stephen还原所使用的锡盐可以通过生成氧化锡沉淀的形态回收,从而减少了对环境的污染。2,3,5,6-四氟苯甲醛在甲醇中用硼氢化钠或硼氢化钾还原得到四氟苄醇。硼氢化钠将醛转化为醇时硼氢化钠用量较少,因而该工艺具有较强的成本优势。
[0027]
本发明提出的一种高效的2,3,5,6-四氟苄醇的制备工艺起始物料易得,发明工艺具有步骤短、硼氢化钠耗用量最少、环境污染小、没有高风险反应的显著优势。
具体实施方式
[0028]
实施例1
[0029]
向3l反应釜中加入2000g 85%甲酸和100g五氟苯腈(0.518mol)。开动搅拌加入539g二水甲酸钠。加热至50-60℃加入6.3g5%pd/c(干基);升温至70-80℃反应5小时。当原料剩余不过1.0%时为反应终点。降温至20-30℃,过滤。滤液水泵减压浓缩。浓缩至干,向釜内加入500g水搅拌。水相用400g二氯甲烷提取2次。有机相常压回收二氯甲烷,减压蒸馏得到78.9g 2,3,5,6-四氟苯腈,收率87%,纯度99.4%。
[0030]
实施例2
[0031]
向3l反应釜中加入2000g 85%甲酸和100g五氟苯腈(0.518mol)。开动搅拌加入539g二水甲酸钠。加热至50-60℃加入3.2g 10%pd/c(干基);升温至70-80℃反应6小时。当原料剩余不过1.0%时为反应终点。降温至20-30℃,过滤。滤液水泵减压浓缩。浓缩至干,向釜内加入500g水,搅拌。水相用400g二氯甲烷提取2次。有机相常压回收二氯甲烷,减压蒸馏得到77.1g 2,3,5,6-四氟苯腈,收率85%,纯度99.0%。
[0032]
实施例3
[0033]
向干燥反应釜中加入250g 2-甲基四氢呋喃,开动搅拌。冰水降温至0~10℃通入干燥氯化氢气体。用湿润ph试纸检测瓶口,当ph试纸迅速变红停止通入氯化氢。加入81.2g二氯亚锡(0.428mol)后搅拌1小时。分批加入50g 2,3,5,6-四氟苯腈(0.2856mol),保温反应5小时。当原料剩余不过1.0%时为反应终点。加水水解。有机相用水洗涤。有机相常压回收2-甲基四氢呋喃后减压蒸馏得到48.2g 2,3,5,6-四氟苯甲醛,纯度98.3%,收率94.7%。
[0034]
实施例4
[0035]
向干燥的反应釜中加入400g叔丁基甲醚,开动搅拌。冰水降温至-10~10℃通入干
燥氯化氢气体。用湿润ph试纸检测瓶口,当ph试纸迅速变红停止通入氯化氢。加入81.2g二氯亚锡(0.428mol)后搅拌1小时。分批加入50g 2,3,5,6-四氟苯腈(0.2856mol),保温反应5小时。当原料剩余不过1.0%时为反应终点。加水水解。有机相用水洗涤。有机相常压回收叔丁基甲醚后减压蒸馏得到46.3g 2,3,5,6-四氟苯甲醛,纯度98.8%,收率91%。
[0036]
实施例5
[0037]
向1l反应釜中加入400g甲醇,开动搅拌。加入200g 2,3,5,6-四氟苯甲醛(1.123mol)。控温于30-40℃分批加入17.85g硼氢化钠(0.47mol)。硼氢化钠加毕,继续搅拌反应1小时。检测反应液,当原料剩余1.0%时反应结束。减压回收甲醇后,降温至15-25℃加入盐酸调ph=3-4。当物料成糊状且内温35-40℃,停止蒸馏。向反应釜中加入200g水。降温至10-20℃,加入400g二氯甲烷提取。静置分层,有机相常压回收二氯甲烷,减压蒸馏得到184g 2,3,5,6-四氟苄醇,收率91.0%,纯度99.2%。
[0038]
实施例6
[0039]
向1l反应釜中加入400g甲醇,开动搅拌。加入200g 2,3,5,6-四氟苯甲醛(1.123mol)。控温于30-40℃分批加入32.4g硼氢化钾(0.6mol)。硼氢化钾加毕,继续搅拌反应1小时。检测反应液,当原料剩余1.0%时反应结束。减压回收甲醇后,降温至15~25℃加入盐酸使ph=3-4。当物料成糊状且内温35-40℃,停止蒸馏。向反应釜中加入200g水。降温至10-20℃,加入400g二氯甲烷提取。静置分层。有机相常压回收二氯甲烷,减压蒸馏得到183g 2,3,5,6-四氟苄醇,收率90.0%,纯度99.3%。
[0040]
实施例7
[0041]
向3000l反应釜中加入1800kg 85%甲酸和100kg五氟苯腈(518mol)。开动搅拌加入539kg二水甲酸钠。加热至50-60℃加入6.3kg 5%pd/c(干基)。升温至70-80℃反应5小时。当原料剩余不过1.0%时为反应终点。降温至20-30℃,过滤。滤液水泵减压浓缩。浓缩至干,向釜内加入500kg水,搅拌。水相用400kg二氯甲烷提取2次。有机相常压回收二氯甲烷,减压蒸馏得到80.1kg 2,3,5,6-四氟苯腈,收率88.3%,纯度99.3%。
[0042]
实施例8
[0043]
向干燥反应釜中加入220kg 2-甲基四氢呋喃,开动搅拌。冰水降温至0-10℃通入干燥氯化氢气体。用湿润ph试纸检测瓶口,当ph试纸迅速变红停止通入氯化氢。加入81.2kg二氯亚锡(428mol)后搅拌1小时。分批加入50kg 2,3,5,6-四氟苯腈(285.6mol),保温反应5小时。当原料剩余不过1.0%时为反应终点。加水水解,有机相用水洗涤。有机相常压回收2-甲基四氢呋喃后减压蒸馏得到48.6kg 2,3,5,6-四氟苯甲醛。纯度98.3%,收率95.5%。
[0044]
实施例9
[0045]
向2000l反应釜中加入780kg甲醇,开动搅拌。加入400kg 2,3,5,6-四氟苯甲醛(2.246kmol)。控温于30-40℃分批加入64.8kg硼氢化钾(1.2kmol)。硼氢化钾加毕,继续搅拌反应2小时。检测反应液,当原料剩余1.0%时反应结束。减压回收甲醇后,降温至15-25℃加入盐酸使ph=3-4。当物料成糊状且内温35-40℃停止蒸馏。向反应釜中加入380kg水。降温至10-20℃,加入800kg二氯甲烷提取。静置分层,有机相常压回收二氯甲烷,减压蒸馏得到369.5kg 2,3,5,6-四氟苄醇,收率90.9%,纯度99.2%。
[0046]
以上实施例描述了本发明的基本原理、主要特征及优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原
理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。