1.本发明属于吸附分离材料技术领域,具体涉及一种铯离子吸附剂及其制备方法和应用。
背景技术:
2.铯是重要的战略资源,由于其特殊的物理化学性质,目前已广泛应用在能源、医药、航空航天、催化、光电通信和油气钻探等高科技领域。
3.我国在青海、西藏和四川等地区的盐湖卤水中储备着丰富的金属资源,其中就包括铯。尽管盐湖卤水中铯储存总量较高,但是多以离子形式存在(cs
),浓度低(小于40mg l-1
),且含有多种伴生离子,特别是物理和化学性质与cs
十分接近的钾离子,分离提取难度极高,目前还没有成功工业化的盐湖卤水铯分离提取工艺技术。
4.现有技术中多采用萃取法和吸附法对铯离子进行吸附分离。萃取法中使用的萃取剂如冠醚类和取代酚类都含有对铯亲和力较强的含氧官能团,如醚氧基和酚羟基,通过多配位作用结合cs
实现极高选择性的分离。例如cn106435180a公开了一种铷离子和铯离子的萃取方法,所述萃取方法包括利用复合萃取剂对含铷离子和铯离子的混合溶液进行萃取处理,所述复合萃取剂包括酸性萃取剂和中性有机物中的至少一种组分以及苯酚类萃取剂;采用上述萃取方法提高了对rb
和cs
的萃取效果。但是,萃取剂不易合成、价格昂贵、容易流失、需要使用有机溶剂,不环保,并且萃取过程需要强酸或强碱等,不适用于盐湖极低浓度cs
的分离提取。
5.吸附法是最适用于低浓度体系的分离方法,主要有无机吸附剂和有机吸附剂两类。例如cn104692406a公开了一种从盐湖卤水中选择性分离铯离子的吸附剂的制备方法,包括:以铯离子为模板剂,按一定配比将表面活性剂、可溶性铯盐、水玻璃、铝酸钠混合,制备得到具有铯离子定制化孔道结构的人工沸石离子筛,对盐湖卤水中cs
选择性吸附性能优异。cn105363414a公开了一种铯离子吸附剂,所述吸附剂的制备原料包括氨基化四氧化三铁、羧基化冠醚衍生物与缩合剂。所述铯离子吸附剂兼具较强的铯离子吸附选择性以及磁性,同时在多次吸附-脱附的循环使用中保持稳定的吸附性能。但是所述吸附剂的制备方法复杂,成本较高。
6.现有技术中普遍缺陷为无机吸附剂部分材料不稳定、容易溶胶化、普遍难解吸,研究多集中在强辐射环境的乏燃料体系。有机吸附剂不耐辐射,选择性有待提高,且制备方法复杂,成本高。
7.因此,开发一种对铯离子选择性高、吸附量高、解吸率高、循环性能好且制备方法简单的吸附剂,是本领域亟待解决的问题。
技术实现要素:
8.针对现有技术存在的不足,本发明的目的在于提供一种铯离子吸附剂及其制备方法和应用。所述铯离子吸附剂通过含酚羟基的单体自交联形成超交联聚合物,对铯离子具
有高选择性、高吸附量以及优异的解吸和循环性能。
9.为达此目的,本发明采用以下技术方案:
10.第一方面,本发明提供一种铯离子吸附剂,所述铯离子吸附剂的原料包括含酚羟基的单体;所述含酚羟基的单体具有式i所示结构;
[0011][0012]
其中,r为-oh或卤素基团中的任意一种。
[0013]
本发明中,所述式i中虚线表示羟基连接在苯环上任意位置。
[0014]
本发明中,所述“卤素”指f、cl、br、i。
[0015]
本发明中,通过选用含酚羟基的单体为原料,使得所述吸附剂富含酚羟基,能够与铯离子进行配位,从而能够吸附分离铯离子;而采用特定含酚羟基的单体能够实现对铯离子的高选择性、高吸附量且具有优异的解吸性能和循环性能。
[0016]
作为本发明优选的技术方案,所述含酚羟基的单体包括对羟基苯甲醇、邻羟基苯甲醇或间羟基苯甲醇中的任意一种或至少两种的组合。
[0017]
优选地,所述含酚羟基的单体包括邻羟基苯甲醇。
[0018]
本发明中,所述含酚羟基的单体选用邻羟基苯甲醇时,对铯离子的选择性更高。
[0019]
优选地,所述铯离子吸附剂的孔径为2~4nm,例如可以为2.1nm、2.2nm、2.3nm、2.4nm、2.5nm、2.6nm、2.7nm、2.8nm、3nm、3.1nm、3.2nm、3.4nm、3.6nm、3.8nm、3.9nm等。
[0020]
本发明中,所述铯离子吸附剂的孔径采用物理吸附仪进行表征。
[0021]
第二方面,本发明提供一种根据第一方面所述的铯离子吸附剂的制备方法,所述制备方法包括:
[0022]
含酚羟基的单体进行傅克烷基化反应,得到所述铯离子吸附剂。
[0023]
优选地,所述反应在催化剂的存在下进行。
[0024]
优选地,所述催化剂与含酚羟基的单体的摩尔比为(1~2):1,例如可以为1:1、1:1.5、1:1.8、1:2等。
[0025]
本发明中,所述催化剂与单体在特定的摩尔比内,所述铯离子吸附剂对铯离子的选择性和吸附量高,催化剂用量太低时,材料聚合困难,产率低;用量太高时,会导致材料交联度过高,疏水性增强,不利于吸附质的扩散,造成吸附性能的大幅下降。
[0026]
优选地,所述催化剂包括路易斯酸催化剂。
[0027]
优选地,所述路易斯酸催化剂包括无水氯化铁、无水氯化铝、无水氯化锡或无水氯化锌中的任意一种或至少两种的组合。
[0028]
本发明中,在路易斯酸的催化下,所述含酚羟基的单体经傅克烷基化反应,能够一步法自交联合成超交联聚合物,弥补现有技术中萃取剂和吸附剂的不足,并兼具多配位萃取剂的高选择性和吸附剂的高吸附量、快速的吸附速率以及优异的解吸和循环性能,同时避免了操作繁琐的预交联步骤和外交联剂的使用,简化了合成过程,降低了成本。
[0029]
优选地,所述反应在溶剂中进行。
[0030]
优选地,所述溶剂包括二氯乙烷。
[0031]
本发明中,以含酚羟基的单体的质量为1g计,所述溶剂的体积为15~25ml,例如可以为16ml、18ml、20ml、22ml、24ml等。
[0032]
优选地,所述反应在保护气氛存在下进行。
[0033]
优选地,所述保护气氛包括氮气和/或氩气。
[0034]
优选地,所述反应包括第一阶段反应和第二阶段反应。
[0035]
优选地,所述第一阶段反应的温度为40~50℃,例如可以为42℃、44℃、45℃、46℃、48℃等。
[0036]
优选地,所述第一阶段反应的时间为4~6h,例如可以为4.5h、5h、5.5h等。
[0037]
优选地,所述第二阶段反应的温度为75~85℃,例如可以为76℃、78℃、80℃、82℃、84℃等。
[0038]
优选地,所述第二阶段反应的时间为15~25h,例如可以为16h、18h、20h、22h、24h等。
[0039]
本发明中,所述反应后还包括抽滤、洗涤和干燥的步骤。
[0040]
优选地,所述洗涤包括依次采用盐酸、超纯水和甲醇进行洗涤的步骤。
[0041]
优选地,所述盐酸的浓度为0.04~0.06mol/l,例如可以为0.045mol/l、0.05mol/l、0.055mol/l等。
[0042]
优选地,所述采用盐酸进行洗涤的时间为1~3h,例如可以为1.5h、2h、2.5h等。
[0043]
优选地,所述采用超纯水洗涤至滤液为中性。
[0044]
优选地,所述采用甲醇洗涤至滤液变为澄清。
[0045]
优选地,所述采用甲醇洗涤后还包括进行索氏提取的步骤。
[0046]
优选地,所述索氏提取的溶剂包括甲醇。
[0047]
优选地,所述索氏提取的时间为20~28h,例如可以为22h、24h、26h、27h等。
[0048]
本发明中,所述索氏提取的目的是除去残留的溶剂和催化剂。
[0049]
优选地,所述干燥的温度为55~65℃,例如可以为56℃、58℃、60℃、62℃、64℃等。
[0050]
优选地,所述干燥的时间为20~28h,例如可以为22h、24h、26h、27h等。
[0051]
优选地,所述干燥在真空干燥箱中进行。
[0052]
作为本发明优选的技术方案,所述制备方法包括以下步骤:
[0053]
在保护气氛存在下,将含酚羟基的单体与催化剂、溶剂混合,在40~50℃的条件下反应4~6h后,在75~85℃的条件下反应15~25h,得到所述铯离子吸附剂。
[0054]
第三方面,本发明提供一种根据第一方面所述的铯离子吸附剂在选择性吸附分离铯离子中的应用。
[0055]
第四方面,本发明提供一种铯离子的吸附分离方法,所述吸附分离方法包括以下步骤:
[0056]
将如第一方面所述的铯离子吸附剂与氯化铯溶液混合,进行吸附分离。
[0057]
优选地,所述混合的金属盐溶液还包括氯化钾、氯化铷或氯化镁中的任意一种或至少两种的组合。
[0058]
优选地,所述混合在碱性条件下进行。
[0059]
优选地,所述混合的溶液中氢氧根离子的浓度为0~0.1mol/l,例如可以为0.01mol/l、0.02mol/l、0.04mol/l、0.05mol/l、0.06mol/l、0.07mol/l、0.08mol/l、
0.09mol/l、0.1mol/l等,进一步优选为0.01~0.05mol/l。
[0060]
本发明中,所述碱性条件通过加入氢氧化钠实现,当naoh浓度为0mol l-1
时,也就是中性条件下,所有的吸附剂几乎不吸附cs
,这是因为酚羟基的pka值为9.99,在中性条件下很难去质子化,需要加入碱提高溶液的ph来推动酚羟基去质子化。其中,最佳naoh浓度为0.025mol l-1
。
[0061]
优选地,所述混合的时间为20~26h,例如可以为22h、24h、25h等。
[0062]
优选地,所述氯化铯与铯离子吸附剂的质量比为(0.002~0.34):1,例如可以为0.004:1、0.008:1、0.01:1、0.015:1、0.02:1、0.05:1、0.1:1、0.15:1、0.2:1、0.25:1、0.3:1等。
[0063]
本发明中,所述混合在恒温旋转摇床中进行。
[0064]
优选地,所述恒温旋转摇床的温度为24~26℃,例如可以为24℃、25℃、26℃等。
[0065]
优选地,所述恒温旋转摇床的转速为150~250rpm,例如可以为160rpm、180rpm、200rpm、220rpm、240rpm等。
[0066]
本发明所述的数值范围不仅包括上述列举的点值,还包括没有列举出的上述数值范围之间的任意的点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0067]
与现有技术相比,本发明的有益效果为:
[0068]
本发明提供的铯离子吸附剂,通过特定含酚羟基的单体自交联形成超交联聚合物,对铯离子具有高选择性、高吸附量以及优异的解吸和循环性能;所述吸附剂对铯离子和钾离子的分离因子≥6.8,对铯离子的吸附量≥229.5mg/g,解吸率≥90.2%,循环保持率≥87.3%。
附图说明
[0069]
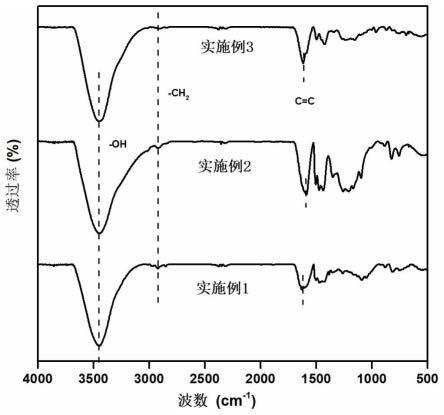
图1为本发明实施例1~3提供的铯离子吸附剂的红外光谱图;
[0070]
图2为本发明实施例1~3提供的铯离子吸附剂的扫描电镜图;
[0071]
其中,a为实施例1提供的铯离子吸附剂的扫描电镜图,b为实施例2提供的铯离子吸附剂的扫描电镜图,c为实施例3提供的铯离子吸附剂的扫描电镜图。
具体实施方式
[0072]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0073]
实施例1
[0074]
本实施例提供一种铯离子吸附剂,所述铯离子吸附剂的原料为对羟基苯甲醇,所述铯离子吸附剂的孔径为2.38nm。
[0075]
本实施例提供一种所述铯离子吸附剂的制备方法,具体步骤包括:
[0076]
在氮气氛围下,将对羟基苯甲醇(2.48g,0.02mol)溶解于50ml二氯乙烷中,加入无水三氯化铁(3.25g,0.02mol)在45℃的条件下反应5h,然后将反应温度升至80℃反应19h;反应结束后,冷却至室温,抽滤,得到不溶性的黑色固体;将所述固体依次采用0.05mol l-1
的盐酸溶液洗涤2h、采用超纯水洗涤至滤液为中性、采用甲醇洗涤至滤液变澄清,随后将分
离出的固体产物进一步用甲醇索氏提取24h,除去残留的溶剂和催化剂,最后在60℃的条件下真空干燥24h,研磨成细粉末,得到所述铯离子吸附剂(hcp-pp)。
[0077]
实施例2
[0078]
本实施例提供一种铯离子吸附剂,所述铯离子吸附剂的原料为邻羟基苯甲醇,所述铯离子吸附剂的孔径为3.19nm。
[0079]
本实施例提供一种所述铯离子吸附剂的制备方法,具体步骤包括:
[0080]
在氮气氛围下,将邻羟基苯甲醇(2.48g,0.02mol)溶解于50ml二氯乙烷中,加入无水三氯化铁(3.25g,0.02mol)在45℃的条件下反应5h,然后将反应温度升至80℃反应19h;反应结束后,冷却至室温,抽滤,得到不溶性的黑色固体;将所述固体依次采用0.05mol l-1
的盐酸溶液洗涤2h、采用超纯水洗涤至滤液为中性、采用甲醇洗涤至滤液变澄清,随后将分离出的固体产物进一步用甲醇索氏提取24h,除去残留的溶剂和催化剂,最后在60℃的条件下真空干燥24h,研磨成细粉末,得到所述铯离子吸附剂(hcp-cp)。
[0081]
实施例3
[0082]
本实施例提供一种铯离子吸附剂,所述铯离子吸附剂的原料为间羟基苯甲醇,所述铯离子吸附剂的孔径为3.91nm。
[0083]
本实施例提供一种所述铯离子吸附剂的制备方法,具体步骤包括:
[0084]
在氮气氛围下,将间羟基苯甲醇(2.48g,0.02mol)溶解于50ml二氯乙烷中,加入无水三氯化铁(3.25g,0.02mol)在45℃的条件下反应5h,然后将反应温度升至80℃反应19h;反应结束后,冷却至室温,抽滤,得到不溶性的黑色固体;将所述固体依次采用0.05mol l-1
的盐酸溶液洗涤2h、采用超纯水洗涤至滤液为中性、采用甲醇洗涤至滤液变澄清,随后将分离出的固体产物进一步用甲醇索氏提取24h,除去残留的溶剂和催化剂,最后在60℃的条件下真空干燥24h,研磨成细粉末,得到所述铯离子吸附剂(hcp-rp)。
[0085]
采用红外光谱仪(德国bruker,tensor 27)对实施例1~3提供的铯离子吸附剂的结构进行表征,结果如图1所示,在1625cm-1
到1591cm-1
附近观察到的特征峰对应于苯环骨架的c=c伸缩振动峰;3446cm-1
处的特征峰对应于-oh的伸缩振动峰;证明聚合后苯酚单体的结构被完全保留下来;3种吸附剂在2920cm-1
至2912cm-1
范围内均出现了一个新特征峰,属于亚甲基的伸缩振动峰,证明成功发生交联反应。
[0086]
采用扫描电子显微镜(英国蔡司,sigma 300)对实施例1~3提供的铯离子吸附剂的形貌进行表征,结果如图2所示,hcp-pp和hcp-cp以不规则的球形颗粒为主,且表面粗糙,而hcp-rp则为不规则块状固体。hcp-pp的颗粒粒径明显较小,属于纳米级颗粒,而hcp-cp的颗粒较大,直径在微米级别。
[0087]
采用物理吸附仪对实施例1~3提供的铯离子吸附剂的孔径进行表征。
[0088]
实施例4
[0089]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,所述制备方法中无水三氯化铁的物质的量为0.04mol,其它步骤及参数均与实施例2相同。
[0090]
实施例5
[0091]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,所述制备方法中无水三氯化铁的物质的量为0.08mol,其它步骤及参数均与实施例2相同。
[0092]
实施例6
[0093]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,将所述邻羟基苯甲醇替换为等摩尔量的对羟基苄基溴。
[0094]
本实施例提供一种铯离子吸附剂的制备方法,具体步骤与实施例2相同。
[0095]
实施例7
[0096]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,将所述邻羟基苯甲醇替换为等摩尔量的邻羟基苄基溴。
[0097]
本实施例提供一种铯离子吸附剂的制备方法,具体步骤与实施例2相同。
[0098]
实施例8
[0099]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,将所述邻羟基苯甲醇替换为等摩尔量的间羟基苄基溴。
[0100]
本实施例提供一种铯离子吸附剂的制备方法,具体步骤与实施例2相同。
[0101]
实施例9
[0102]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,将所述邻羟基苯甲醇替换为等摩尔量的对羟基苄基氯。
[0103]
本实施例提供一种铯离子吸附剂的制备方法,具体步骤与实施例2相同。
[0104]
实施例10
[0105]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,将所述邻羟基苯甲醇替换为等摩尔量的邻羟基苄基氯。
[0106]
本实施例提供一种铯离子吸附剂的制备方法,具体步骤与实施例2相同。
[0107]
实施例11
[0108]
本实施例提供一种铯离子吸附剂,其与实施例2的区别仅在于,将所述邻羟基苯甲醇替换为等摩尔量的间羟基苄基氯。
[0109]
本实施例提供一种铯离子吸附剂的制备方法,具体步骤与实施例2相同。
[0110]
实验例
[0111]
分别配制0mol l-1
、0.005mol l-1
、0.02mol l-1
、0.05mol l-1
、0.1mol l-1
、0.2mol l-1
的naoh溶液和10.0mmol l-1
的cscl溶液,称取0.01g实施例1~3提供的铯离子吸附剂于10ml离心管中,加入2ml naoh溶液和2ml cscl溶液,放置于恒温旋转摇床(25℃,200rpm)中恒温震荡24小时后使其达到吸附平衡,高速离心后用注射器取上清液,用0.22μm的水系滤头过滤后稀释20倍,使用电感耦合等离子发射光谱仪(icp-aes)测定溶液中铯离子浓度,计算铯离子的吸附量,吸附量其中,c0(mg l-1
)是初始金属离子的浓度,ce(mg l-1
)是吸附平衡时溶液中金属离子的浓度,v(l)是溶液体积,m(g)是加入吸附剂的质量;结果如表1所示。
[0112]
表1
[0113][0114]
由表1可知,随着naoh浓度的增加,所述吸附剂对cs
的吸附量呈现一个先迅速增加后减少的趋势,最终溶液中naoh浓度最优为0.025mol l-1
,因此,下文关于性能测试中氢氧化钠最终浓度为0.025mol l-1
。
[0115]
性能测试
[0116]
(1)选择性:称取0.01g实施例1~11提供的铯离子吸附剂于10ml离心管中,加入2ml、0.05mol l-1
的naoh溶液和2ml含干扰离子的氯化铯溶液,放置于恒温旋转摇床(25℃,200rpm)中恒温震荡24小时后使其达到吸附平衡,高速离心后用注射器取上清液,用0.22μm的水系滤头过滤后稀释20倍,使用icp-aes测定溶液中铯离子浓度,计算分离因子;
[0117]
平衡分配系数分离因子
[0118]
其中,c0(mg l-1
)是初始金属离子的浓度,ce(mg l-1
)是吸附平衡时溶液中金属离子的浓度,v(l)是溶液体积,m(g)是加入吸附剂的质量;为铯离子的平衡分配系数,为干扰离子的平衡分配系数;
[0119]
本发明中,干扰离子为钾离子时,配制钾铯离子摩尔比为分别为14:1、69:1、357:1、684:1、1358:1的混合溶液,计算5种浓度下分离因子的平均值;
[0120]
干扰离子为铷离子时,铷铯离子的摩尔比为15:1;
[0121]
干扰离子为镁离子是,镁铯离子摩尔比为467:1。
[0122]
(2)吸附量:分别配制10mg l-1
、50mg l-1
、100mg l-1
、550mg l-1
、630mg l-1
、720mg l-1
、870mg l-1
、1270mg l-1
、1700mg l-1
的cscl溶液;称取0.01g实施例1~11提供的铯离子吸附剂于10ml离心管中,加入2ml naoh溶液和2ml cscl溶液,放置于恒温旋转摇床中(25℃,200rpm)恒温震荡24小时后使其达到吸附平衡,高速离心后用注射器取上清液,用0.22μm的水系滤头过滤后稀释20倍,使用icp-aes测定溶液中铯离子浓度;
[0123]
采用langmuir吸附等温模型计算最大吸附量的表达方程式为:
[0124][0125]
其中,ce(mg l-1
)是吸附平衡时溶液中吸附质的浓度,qe(mg g-1
)是吸附平衡时的吸附容量,qm(mg g-1
)是吸附剂的最大吸附容量,k
l
(l mg-1
)是langmuir模型常数。
[0126]
(3)解吸和循环性能:称取0.01g实施例1~11提供的铯离子吸附剂于10ml离心管中,加入2ml naoh溶液和2ml 10mmol l-1
的cscl溶液,放置于恒温旋转摇床(25℃,200rpm)中恒温震荡24小时后使其达到吸附平衡,高速离心后用注射器吸出上清液,用0.22μm的水系滤头过滤后稀释20倍,使用icp-aes测定溶液中铯离子浓度,计算吸附量;在剩余吸附剂中加入4ml 0.05mol l-1
的hcl在与吸附同等条件下解吸1小时,高速离心后用注射器吸出上清液,用0.22μm的水系滤头过滤后稀释20倍,使用icp-aes测定溶液中铯离子浓度,计算解吸率;随后吸附剂反复洗至中性后再继续下一次吸附-解吸实验,连续循环5次;计算5次循环的平均解吸率;
[0127]
解吸率=单位质量吸附剂解吸的吸附质质量/单位质量吸附剂吸附的吸附质质量
×
100%;
[0128]
循环保持率=第一次循环吸附剂的吸附容量/第五次循环吸附剂的吸附容量
×
100%。
[0129]
具体测试结果如表2所示:
[0130]
表2
[0131]
[0132]
由上表可知,本发明提供的铯离子吸附剂,通过选用特定含酚羟基的单体形成超交联多孔吸附剂,对铯离子具有高选择性和高吸附量,且解吸速率快、效率高,循环性能好。由实施例1~4可知,所述铯离子吸附剂对铯钾离子的分离因子为13.6~44.7,对铯铷离子的分离因子为6.3~13.8,对铯镁的分离因子为2.9~10.1,对铯离子具有高选择性;对铯离子的最大吸附量为229.5~302.1mg/g,解吸率为90.4~93.8%,循环保持率为87.36~91.3%。
[0133]
由实施例2与实施例1和3比较可知,当选用邻羟基苯甲醇时,吸附剂对铯离子的吸附分离效果更好;通过实施例2与实施例5比较可知,所述催化剂与单体的质量比不再特定的范围内,对铯离子的选择性变差,且吸附量降低;由实施例2与实施例6~11比较可知,并非本发明特定的单体时,所述吸附剂对铯离子的选择性变差。
[0134]
综上所述,本发明提供的铯离子吸附剂通过选用特定含酚羟基的单体,自交联形成多孔有机吸附剂,既实现对铯离子的高选择性,又能实现对铯离子的高吸附量,且具有优异的解吸循环性能,避免外加交联剂,制备方法简单,节约成本且环保。
[0135]
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。