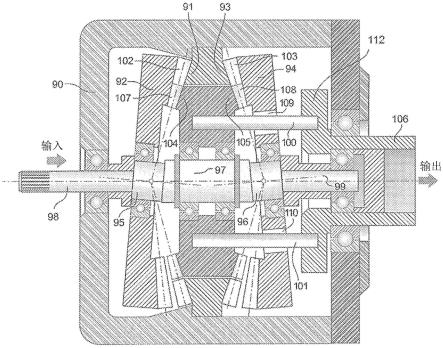
通过靶向氨酰基脯氨酸二肽酶(pepd)重新激活p53突变体用于癌症治疗
1.相关申请的交叉引用
2.本技术要求于2019年8月29日提交的美国临时专利申请号62/893,367的优先权,其全部公开内容通过引用并入本文。
3.关于联邦资助的研究或开发的声明
4.本发明是在由美国国立卫生研究院(national institutes of health)授予的资助号ca215093和ca164574下的政府资助下完成的。政府对本发明享有一定的权利。
背景技术:
5.p53肿瘤抑制因子在抑制癌症发生和进展中起着至关重要的作用,但通常在所有类型的人类癌症中都突变。有许多p53突变体,其中绝大多数是dna结合域中的单氨基酸变化。突变p53失去其肿瘤抑制活性或获得致癌活性。对用于治疗表达这样的p53突变体的癌症的组合物和方法存在持续且未满足的需求。本公开与此需求相关。
技术实现要素:
6.本公开提供了用于预防和/或治疗癌症的组合物和方法。这些方法通常涉及靶向表达促进癌细胞增殖的p53突变形式的癌细胞中的肽酶d(pepd),也称为氨酰基脯氨酸二肽酶。
7.在得出目前提供的公开内容时,分析了pepd是否结合并调节p53突变体,因为负责pepd结合的p53结构域(prd)在几乎所有癌症相关的p53突变体中都是完整的。我们最近发现,pepd结合并抑制野生型(正常)p53,并且通过靶向pepd来破坏缔合会释放并激活p53,从而导致细胞死亡。然而,一个主要的担忧是,如果pepd确实与p53突变体结合,则靶向pepd可能会释放p53突变体并解放它们的显性负效应或致癌效应,即促进癌细胞生长和增殖。
8.本公开包括对常见癌症相关p53突变体(包括r175h、r248q、r273h、r280k和e285a)的评估。众所周知,这些突变体已失去肿瘤抑制功能并获得显性负功能或致癌功能。然而,令我们惊讶的是,通过sirna的pepd敲低(kd)并没有释放p53突变体以促进细胞存活和生长,而是导致表达p53突变体的癌细胞死亡并引起指示wt-p53活性的分子变化。除了下面描述的某些例外情况外,这种现象不仅限于特定的突变体。pepd缺失引起的细胞死亡显然是由p53突变体引起的,因为敲除突变体会使细胞对pepd抑制不敏感。这些发现挑战了当前对p53突变体生物学的理解,揭示了p53突变体的关键调控机制,因此提供了一种重新激活p53突变体的新策略,该策略可能适用于大多数(如果不是全部)可能有或可能没有致癌作用的p53突变体。在实施方案中,本公开因此包括抑制包含p53突变体的癌细胞中pepd的表达。本公开在使用rna抑制和pepd敲除的非限制性实施方案中进行了说明。结果包括在相关动物模型中证明了pepd敲低对同基因肿瘤(无论是否表达p53突变体)的生长和其中关键蛋白质的表达的体内影响。因此,在实施方案中,本公开包括引入到包含p53突变体的癌细胞,所述p53突变体可以是功能丧失p53突变体、显性负p53突变体和功能获得p53突变体。
附图说明
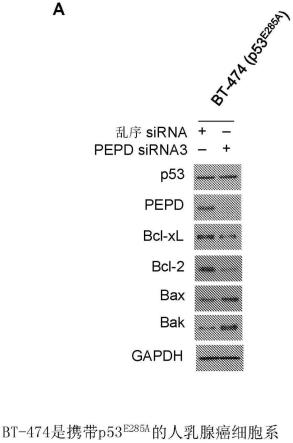
9.图1.pepd敲低重新激活p53突变体。a,细胞用sirna(10nm)处理48小时,从中制备全细胞裂解物并通过蛋白质印迹(wb)进行分析。甘油醛3-磷酸脱氢酶(gapdh)是上样对照。b,细胞用sirna(10nm)处理72小时,然后通过台盼蓝活力测定法进行分析(平均值
±
sd,n=3)。*p《0.0001。
10.图2.通过sirna的pepd kd可诱导p53
e285a
突变体的转录非依赖性肿瘤抑制活性。a.sirna(10nm)处理72小时后细胞中p53重定位的wb分析。测量核纤层蛋白a、α-微管蛋白和电压依赖性阴离子通道(vdac)以排除亚细胞级分的交叉污染或作为上样对照。b,用sirna(10nm)处理细胞72小时后p53与线粒体中亲环蛋白d(cypd)结合的wb分析。c,用sirna(10nm)处理细胞72小时后线粒体膜电位(mmp)的流式细胞术分析。误差线是从3个实验计算的平均值
±
sd。
11.图3.在bt-474细胞中pepd kd诱导p53突变体的磷酸化和转录活性。a,质粒转染24小时,然后用sirna(10nm)处理48小时后细胞中的报告子活性。pg13-luc是包含多个拷贝的p53
wt
结合位点的p53
wt
报告子,而mg15-luc包含多个拷贝的突变p53结合位点而对p53
wt
无响应。用pg13-luc或mg15-luc以及用于控制转染效率的prl-tk转染细胞。b和c,用sirna(10nm)处理72小时的细胞中p53
e285a
的磷酸化的phostag-wb和wb分析。d,用sirna(10nm)处理72小时的细胞的核级分和胞质溶胶的wb分析和phostag-wb分析。测量核纤层蛋白a和gapdh作为上样对照。
12.图4.bt-474细胞中pepd与p53突变体的结合以及pepd kd的影响。a-d,与pepd结合的细胞p53
e285a
和与p53
e285a
结合的细胞pepd的百分比的免疫沉淀(ip)-wb分析。ib条带由imagej量化。误差线是平均值
±
sd(n=3)。e,用sirna处理72小时的细胞的核级分中pepd和p53
e285a
的wb分析以及无pepd的p53
e285a
和磷酸化-p53
e285a
的ip-wb分析。测量核纤层蛋白a作为上样对照。
13.图5.通过crisp-cas9的pepd ko以p53依赖性方式杀死人癌细胞。用对照或靶向pepd的crispr-cas9处理细胞72小时,显示了代表性的显微图像(比例尺:100μm)。
14.mda-mb-231(p53
r280k
)是一种人乳腺癌细胞系。mda-mb-231(p53-/-)是通过crispr-cas9基因编辑从mda-mb-231(p53
r280k
)产生的。
15.图6.pepd与p53
wt
和突变体的结合。a.通过酶联免疫吸附测定(elisa)测量的pepd与野生型p53(p53
wt
)和突变体的直接结合。b.未经处理的细胞的全细胞裂解物的wb分析。gapdh是上样对照。c、d.通过ip使样品中的所有pepd或p53沉淀下来(pulled down)。同种型特异性igg用作对照。通过用elisa测量上清液中的p53和pepd来计算不相互结合的p53和pepd的百分比。通过wb分析沉淀物和输入(图14a、14b),通过将相互结合的p53和pepd的条带强度与输入的条带强度(通过imagej测量)进行比较来计算相互结合的p53和pepd的百分比。e.通过elisa测量的全细胞裂解物中的pepd和p53水平。(a)和(c-e)中的平均值
±
sd(n=3)。另请参见图13和14。
16.图7.pepd kd对细胞存活和关键蛋白质水平的影响,以及pepd
g278d
的反作用。a.用sirna(10nm)处理72小时的细胞通过台盼蓝测定法测量的活力。b.全细胞裂解物的wb分析。gapdh是上样对照。细胞用sirna(10nm)处理48小时。c.通过ip使全细胞裂解物中的所有pepd沉淀,并通过wb分析上清液。gapdh是上样对照。p53以两次曝光显示,以清晰显示所有
条带。细胞用sirna(10nm)处理48小时。未经处理的mda-mb-231细胞的裂解物在wb中用作阳性对照。d、e.通过台盼蓝测定法测量的细胞活力,以及全细胞裂解物的wb分析。gapdh是上样对照。将细胞用等量的质粒转染,并且24小时后用载剂或pepd sirna(10nm)处理72小时。(a)和(d)中的平均值
±
sd(n=3)。***p《0.001;****p《0.0001;ns,不显著。另请参见图15。
17.图8.pepd kd对同基因细胞中p53突变体的影响。a.全细胞裂解物的wb分析。gapdh是上样对照。mda-mb-231(p53
ko
)细胞用等量的质粒(空载体或p53突变体)转染48小时。b、c.通过台盼蓝测定法测量的细胞活力,以及全细胞裂解物的wb分析。gapdh是上样对照。mda-mb-231(p53
ko
)细胞用p53突变体(每种突变体等量的质粒)转染,24小时后用sirna(10nm)处理96小时。(b)中的平均值
±
sd(n=3)。****p《0.0001。
18.图9.pepd kd对p53突变体的转录非依赖性肿瘤抑制活性的影响。a.亚细胞级分的wb分析,使用电压依赖性阴离子通道(vdac)、gapdh和核纤层蛋白b作为上样对照。细胞用sirna(10nm)处理48小时。b.通过jc-1荧光测量的线粒体膜电位损失。细胞用sirna(10nm)处理48小时。c.细胞用sirna(10nm)处理48小时,从中分离线粒体并进行ip。通过wb分析免疫沉淀物。d.通过tunel测定法测量的凋亡。细胞用sirna(10nm)处理72小时(参见图15d)。(b)和(d)中的平均值
±
sd(n=3)。另请参见图15和图16。
19.图10.pepd kd对p53突变体的磷酸化和转录活性的影响。a.全细胞裂解物的wb分析。细胞用sirna(10nm)处理24小时。b.亚细胞级分的phostag wb分析。细胞用sirna(10nm)处理48小时。c.在全细胞裂解物中测量的荧光素酶活性。细胞用等量的pg13-luc或mg15-luc以及prl-tk转染,24小时后用sirna(10nm)处理48小时。d.通过chip-qpcr测定法测量的p53突变体与cdkn1a (编码p21)和bbc3(编码puma)的启动子中的p53
wt
结合位点的结合。细胞用sirna(10nm)处理48小时。(c)和(d)中的平均值
±
sd(n=3)。另请参见图17。
20.图11.pepd kd对p53突变体的重新折叠的影响,以及k373乙酰化的作用。a.全细胞裂解物的wb分析。gapdh是上样对照。细胞用sirna(10nm)处理48小时。b.亚细胞级分的wb分析。skbr3细胞用sirna(10nm)处理48小时。c、d.通过台盼蓝测定法测量的细胞活力,以及全细胞裂解物的wb分析。gapdh是上样对照。细胞在具有或不具有c646(8μm)的情况下用sirna(10nm)处理72小时。c646是使p53乙酰化的p300/cbp的抑制剂。e、f.通过台盼蓝测定法测量的细胞活力和全细胞裂解物的wb分析。gapdh是上样对照。将p53
r175h/k373r
转染到mda-mb-231(p53
ko
)细胞中;24小时后,细胞用sirna(10nm)处理96小时。g-k.使用pab1620、pab240或同种型匹配的igg对全细胞裂解物进行p53的ip,并对免疫沉淀物进行wb分析。pab1620和pab240分别检测p53的“野生型”和“变性”构象。(i)中的相对p53
r175h
水平由imagej测量。细胞未经处理(g、h)或在具有或不具有c646(8μm)的情况下用sirna(10nm)处理48小时(i、j)。将p53
r175h/l373r
转染到mda-mb-231(p53
ko
)细胞中;24小时后,细胞用sirna (10nm)处理48小时(k)。l.亚细胞级分的wb分析。将p53
r175h/k373r
转染到mda-mb-231(p53
ko
)细胞中;24小时后,细胞用sirna(10nm)处理48小时。核纤层蛋白b、α-微管蛋白和vdac用作上样对照并排除交叉污染(b、l)。cal-51全细胞裂解物用作wb的p53
wt
输入。(b、e)中的平均值
±
sd(n=3)。*p《0.05;****p《0.0001。另请参见图18。
21.图12.pepd kd对表达或不表达p53突变体的同基因肿瘤的生长或其中关键蛋白的表达的影响。a-f.每3天通过肿瘤内注射sirna(10pmol)治疗具有由mda-mb-231-p53
r280k
、mda-mb-231-p53
ko
或mda-mb-231-p53
r175h
细胞产生的原位乳腺肿瘤的小鼠。箭头表示治疗
开始。实验在最后一次给药后24或48小时结束。平均值
±
sem(n=13-16)。****p《0.0001。g.肿瘤组织匀浆物的wb分析。gapdh是上样对照。对每组两个肿瘤进行分析。h.pepd kd重新激活p53突变体的图形总结。a,乙酰化;p,磷酸化;u,单泛素化;p53m,p53突变体。另请参见图19。
22.图13.pepd、p53
wt
、p53突变体和细胞系的表征,与图6相关。a.经亲和纯化的重组pepd、p53
wt
、p53突变体和银染的十二烷基硫酸钠聚丙烯酰胺凝胶电泳。b.细胞系中p53的sanger测序,重点关注氨基酸#175、#248、#273和#280的变化。c.通过rt-pcr测量细胞中的pepd mrna水平,使用gapdh mrna作为对照。
23.图14.细胞中pepd与p53
wt
及其突变体的结合,与图6相关。a.使用过量的pepd抗体通过ip使样品(全细胞裂解物、胞质溶胶或核提取物)中的所有pepd沉淀下来。通过wb分析沉淀物和输入。用于p53 wb的输入为pepd ip中使用的每个样品的10%、15%或20%,其中skbr3和mda-mb-231的全细胞裂解物为10%输入,skbr3和mda-mb-231的胞质溶胶和核提取物为15%输入,以及cal-51(全细胞裂解物、胞质溶胶和核提取物)为20%输入。还对选择的上清液进行了wb分析,以确认所有pepd都被沉淀下来。进行了三次实验来计算图6c中所示的结合百分比。b.使用过量的p53抗体通过ip使样品中的所有p53沉淀下来。通过wb分析沉淀物和输入。还对选择的上清液进行了wb分析,以确认所有p53都被沉淀下来。用于pepd wb的输入是p53 ip中使用的每个样品的15%。还对选择的上清液进行了wb分析,以确认所有p53都被沉淀下来。进行了三次实验来计算图6d中所示的结合百分比。c.核提取物和胞质溶胶中核纤层蛋白b和α-微管蛋白的wb分析以排除交叉污染。
24.图15.pepd kd对p53和其它蛋白质的水平、细胞周期进程和凋亡的影响,与图7和图9相关。a.使用gapdh作为上样对照,对全细胞裂解物的pepd和其它蛋白质进行wb分析。细胞用sirna(10nm)处理48小时。b.通过流式细胞术测量细胞周期进程。细胞用sirna(10nm)处理48小时。平均值
±
sd(n=3)。**p《0.01;****p《0.0001。c.使用gapdh作为上样对照,对全细胞裂解物的裂解的胱天蛋白酶3进行wb分析。细胞用sirna(10nm)处理48小时。d.用fluor-594进行tunel荧光染色和用4',6-二脒基-2-苯基吲哚(dapi)进行核荧光染色。在染色前细胞用sirna(10nm)处理72小时。比例尺:100μm。
25.图16.mdm2在skbr3细胞中p53
r175h
的线粒体富集中的作用,与图9相关。a.全细胞裂解物、细胞裂解物减去线粒体和线粒体的wb分析。gapdh、vdac和α-微管蛋白用作上样对照以排除交叉污染。细胞用乱序sirna(scramble sirna)或mdm2 sirna(10nm)处理,并且24小时后用乱序sirna或pepd sirna (10nm)处理48小时。b.细胞裂解物减去线粒体的wb分析。gapdh是上样对照。对相同的样品也进行p53
r175h ip,并且通过wb分析免疫沉淀物。细胞用sirna(10nm)处理48小时。c.使用泛素(ub)抗体对线粒体样品和核提取物进行ip,并通过wb分析免疫沉淀物。细胞用sirna(10nm)处理48小时,最后4小时加入泛素醛(100μm),从中分离线粒体。
26.图17.pepd kd对p53
wt
和p53突变体的磷酸化的影响,与图10相关。a.全细胞裂解物的wb分析。细胞用sirna(10nm)处理48小时。b.细胞用sirna(10nm)处理48小时,从中制备核提取物。对提取物进行pepd ip。通过wb和phostag wb分析免疫沉淀物和上清液二者。将用乱序sirna和pepd sirna处理的细胞样品中的p53量平衡以用于phostag wb。
27.图18.pepd kd对p53突变体的再折叠的影响,以及k373乙酰化在p53突变体的再折
叠和重新激活中的作用,与图11相关。a.亚细胞级分(包括核提取物、胞质溶胶和线粒体)的wb分析。mda-mb-231细胞用sirna(10nm)处理48小时。b.用过量的pepd抗体对skbr3细胞和mda-mb231细胞的核提取物进行ip以使所有pepd沉淀下来,然后对沉淀物和上清液二者进行wb分析。细胞用sirna(10nm)处理48小时。c-f.通过台盼蓝测定法测量的细胞活力和全细胞裂解物的wb分析。gapdh是上样对照。mda-mb-231细胞、mda-mb-468细胞和hcc70细胞在具有或不具有c646(8μm)的情况下用sirna(10nm)处理72小时(c、d)。mda-mb-231(p53
ko
)细胞用包括p53
r248q/k373r
、p53
r273h/k373r
和p53
r280k/k373r
的p53突变体转染,24小时后,用sirna (10nm)处理96小时(e、f)。g.用pab1620或pab240对重组p53
wt
和p53
r175h
进行ip,然后对p53
wt
和p53
r175h
的沉淀物进行wb分析。h-k.使用pab1620或pab240对全细胞裂解物进行ip,然后对沉淀物(h-k)和上清液(h)进行wb分析。mda-mb-23细胞、mda-mb-468细胞和hcc70细胞未经处理(h)或在具有或没有8mm c646的情况下用sirna (10nm)处理48小时(i、j)。mda-md-231(p53
ko
)细胞用包括p53
r248q/k373r
、p53
r273h/k373r
和p53
r280k/k373r
的p53突变体转染,24小时后用sirna(10nm)处理48小时(k)。在(h)中使用未经处理的全细胞裂解物作为wb的对照。l.亚细胞级分的wb分析。将p53
r280k/k373r
转染到mda-mb-231(p53
ko
)细胞中;24小时后,细胞用sirna(10nm)处理48小时。核纤层蛋白b、α-微管蛋白和vdac用作上样对照并排除交叉污染(a、l)。(b、e)中的平均值
±
sd(n=3)。*p《0.05;****p《0.0001。
28.图19.稳定表达p53
r175h
的mda-mb-231细胞的表征,与图12相关。a.全细胞裂解物的wb分析。用p53
r175h
转染mda-mb-231(p53
ko
)细胞,并通过嘌呤霉素选择表达突变体的稳定克隆。skbr3细胞用于在wb中进行比较。gapdh是上样对照。b.通过台盼蓝测定法测量的细胞活力。细胞用sirna(10nm)处理72小时。平均值
±
sd(n=3),p《0.0001。
具体实施方式
29.除非本文另有定义,否则本公开中使用的所有技术和科学术语具有与本公开所属领域的普通技术人员通常理解的相同含义。
30.在整个本说明书中给出的每个数值范围包括其上限值和下限值,以及落入其中的每个较窄的数值范围,就好像这些较窄的数值范围都明确地写在本文中一样。
31.本公开包括本文公开的所有多核苷酸、它们的互补序列和反向互补序列。对于通过数据库条目的方式对多核苷酸或氨基酸序列的任何引用,数据库条目中呈现的多核苷酸和氨基酸序列并入本文,如同其在本技术或专利的有效提交日期存在。
32.在实施方案中,本公开包括抑制表达p53突变形式的癌细胞中pepd的表达,该突变形式已失去肿瘤抑制功能、和/或已获得显性负功能、和/或已获得致癌功能。
33.已经描述了抑制癌细胞中pepd的表达。(yang等人nature communications,volume 8,article number:2052(2017))。简单来说,yang等人的参考文献描述了pepd结合并抑制超过一半的核和细胞质p53,而不依赖其酶活性。该参考文献还表明,pepd与p53中的富含脯氨酸结构域(prd)结合,从而抑制核p53的磷酸化和mdm2介导的核和细胞质p53的线粒体易位。该参考文献还表明,消除pepd会导致由于p53激活的细胞死亡和肿瘤消退。yang等人的参考文献使用了表达野生型p53(除了一个突变体外)的细胞。然而,yang等人在那篇论文中得出结论,p53突变体(在um-uc-3细胞中发现的p53的f113c变化)仍然具有功能,因此尽管发生了这种突变,但认为它保留了其野生型功能。因此,yang等人的参考文献没有描
述或暗示将pepd与已知具有致癌作用的p53突变体分离的后果。
34.相比之下,本公开涉及抑制pepd和p53突变形式之间的相互作用,其结果出乎意料地导致癌细胞死亡。因此,本公开的方法是违反直觉的,预期的是从与pepd的复合物中释放已失去其肿瘤抑制功能并且已获得致癌功能的突变p53应该解放其致癌功能并加速肿瘤生长。然而,本公开中提供的数据出人意料地证明了相反的效果,即破坏pepd与突变p53的相互作用可预期诱导癌细胞死亡。
35.因此,在实施方案中,本公开包括在包含除了保留野生型肿瘤抑制功能的突变外的致癌突变p53蛋白的癌细胞中抑制pepd与p53的相互作用。因此,本公开包括,在某些非限制性实施方案中,前提是癌细胞不包含其中唯一突变是f113c的突变p53。
36.在实施方案中,在不存在本文所述的pepd靶向剂的情况下,形成包含突变p53和野生型p53的复合物(例如异二聚体)抑制野生型p53的功能。
37.在实施方案中,癌细胞包含突变p53,其在不存在本文所述的pepd靶向剂的情况下与pepd形成复合物。
38.在实施方案中,抑制pepd和突变p53的复合物的形成会将突变p53转化为模拟野生型p53功能的p53,从而杀死癌细胞,而不是促进肿瘤生长的其它预期结果。因此,本公开包括通过靶向pepd重新激活p53突变体用于癌症治疗。在实施方案中,在施用本文所述的rnai剂之后,最初存在于与突变体p52的复合物中的pepd通过普通的细胞蛋白降解方法自然降解,并且由于rnai剂的存在,不产生替代pepd,从而使突变体p53摆脱pepd,获得由某些翻译后修饰(反式激活结构域中的赖氨酸373乙酰化、单泛素化和磷酸化)诱导的野生型确认,并因此参与杀死癌细胞。在实施方案中,本公开提供了破坏p53突变体与pepd缔合的药剂(例如具有该功能的小分子或其它化合物)的用途。
39.在实施方案中,本公开的药剂所靶向的癌细胞中的突变p53已失去其作为转录调节因子的正常功能、或已失去其正常的转录非依赖性功能,并且异常功能和相关突变是本领域熟知的。
40.在实施方案中,使用本公开的药剂靶向的癌细胞包括功能丧失p53突变体、显性负p53突变体和/或功能获得p53突变体,其中的每一种在本领域中也是众所周知的。
41.在实施方案中,癌细胞表达突变p53和野生型p53,因此治疗的个体可能具有p53等位基因杂合的肿瘤。在实施方案中,癌细胞可能仅表达突变的p53,因此治疗的个体可能是p53等位基因的纯合子。
42.促进癌症生长和表征可以根据本公开靶向的癌细胞的p53突变已详细描述,例如,在freed-pastor and prives,genes&dev(2012)26:1268-1286中,其公开内容通过引用并入本文。
43.p53的氨基酸序列及其伴随的氨基酸位置在本领域中是众所周知的并且由氨基酸位置来指代。在非限制性实例中,癌细胞表达具有位于p53氨基酸r175、r248、r273、r280或e285处的突变的p53。这些位置突变的具体和非限制性实例包括r175h、r248q、r273h、r280k和e285a,并且另外的实例如下所述。在实施方案中,p53突变是以下中的一种或任何组合:k132q、a138p、l145r、v147d、p151s、p152l、g154v、t155p、t155n、r156p、v157f、r158l、r158h、a159d、a161t、y163c、k164n、r175l、c176f、c176y、c176w、h179q、r181c、h193l、h193r、h193y、l194f、r213q、y205f、s215i、v216m、y220c、y220d、p223l、e224d、i232n、
y234h、y236c、m237i、n239d、s241f、c242f、c242r、c242s、c242w、r244d、g245r、g245s、r248w、r248l、g245s、g245c、g245d、g245v、m246i、r249s、p250l、i251n、i254d、i255n、g262v、g262d、g266e、g266v、r267l、r267w、v272m、r273c、r273l、v274f、c275g、c275f、c277f、p278a、r280t、r282w、r282g、r283c、r283p、p309s、k320n、q331r、g334v、g389w。
44.在实施方案中,本公开的方法靶向的癌细胞中的p53突变改变了p53与一种或多种其它蛋白质的相互作用,包括但不一定限于nf-y、sp1、ets-1、vdr、srebp-2、topbpi、pin1、mre11、pml、p63或p73。在实施方案中,这样的p53突变包括上述那些,并且可以进一包括在p53位置v143、d281、r249和y220处的突变。
45.在实施方案中,p53突变体诱导癌基因的表达,其非限制性实例包括增殖细胞核抗原(pcna)、egfr、c-myc和混合谱系白血病1(mll1)。
46.在实施方案中,如本文所述,当通过pepd kd不被靶向时,癌细胞中的p53突变可以反式激活其它蛋白质,包括但不限于myc、cxcl1、pcn、map2k3、ccna、ccnb、cdk1、cdc25c、asns、e2f5、mcm6、igf1r、stmn1和egfr,从而促进癌细胞的增殖。
47.在实施方案中,本文所述的当通过pepd kd不被靶向时,癌细胞中的p53突变体可上调编码抑制细胞凋亡或促进对化学治疗剂抗性的蛋白质的基因。这样的基因包括但不限于egr1、abcb1、igf2、dut、bcl2l1、timm50、lgals3和nfkb2。
48.在实施方案中,用于抑制包含pepd和突变p53的复合物形成的药剂是rnai剂。在实施方案中,rnai剂因此与编码pepd的mrna具有互补性。人pepd和编码它的mrna的序列在本领域中是已知的,并且本公开包括靶向任何这样的mrna序列。例如,参见美国专利号10,155,028,其中pepd的描述通过引用并入本文。
49.因此,在实施方案中,可以通过抑制编码pepd的mrna的翻译来抑制包含如本文所述的突变p53的癌细胞中pepd的表达。在实施方案中,编码蛋白质的mrna被降解。在这方面,在非限制性实施方案中,进行rna干扰(rnai)介导的对编码pepd的mrna的沉默和/或减少。在实施方案中,这通过递送任何合适的rnai剂来实现。在实施方案中,使用基于sirna的方法。这可以通过在细胞中引入和/或表达一种或多种合适的短发夹rna(shrna)来进行。shrna是包含有义链、反义链以及在有义片段和反义片段之间的短环序列的rna分子。shrna被输出到细胞质中,在那里它被dicer加工成短干扰rna(sirna)。sirna是21-23个核苷酸的双链rna分子,可被rna诱导的沉默复合物(risc)识别。一旦整合到risc中,sirna就会促进靶mrna的切割和降解。因此,为了用于如本文所述的rnai介导的pepd表达的沉默或下调,可以使用sirna、shrna或mirna。在替代实施方案中,使用功能性rna,例如核酶。在实施方案中,核酶包括锤头状核酶、发夹状核酶或丁型肝炎病毒核酶。在相关实施方案中,可以使用适合靶向相关mrna的微rna(mirna)。术语“微rna”可以与“mir”或“mirna”互换使用,以指代例如来自工程化mirna基因的未加工或加工的rna转录物。未加工的mirna基因转录物也称为“mirna前体”,通常包含长度为约70-100个核苷酸的rna转录物。mirna前体可以通过用rna酶(例如,dicer、argonaut或rna酶iii)消化而被加工成有活性的19-25个核苷酸的rna分子。这种有活性的19-25个核苷酸的rna分子也被称为“经加工的”mirna基因转录物或“成熟的”mirna。任何这些形式的微rna都可以适用于本公开的实施方案。此外,在某些实施方案中,rnai剂可以作为合成剂提供,例如微rna模拟物、短干扰rna(sirna)、rna干扰(rnai)分子、双链rna(dsrna)、如上所述的短发夹rna(shrna)、初级mirna(pri-mirna)或小核仁
rna(snorna)。因此可以通过抑制翻译、转录和/或通过mrna降解来实现对pepd表达的抑制。
50.在实施方案中,可以修饰rnai剂以改善其功效,例如通过抗核酸酶消化。在实施方案中,rnai剂多核苷酸包含修饰的核糖核苷酸或脱氧核糖核苷酸,因此包括rna/dna杂合体。在非限制性实例中,修饰的核糖核苷酸可以包含用含有1-6个饱和或不饱和碳原子的
‑‑
o-烷基或用具有3-6个碳原子的芳基对核糖部分的2'位置的甲基化和/或取代,其中这样的烷基或芳基可以未被取代或可以被例如卤素、羟基、三氟甲基、氰基、硝基、酰基、酰氧基、烷氧基、羧基、碳烷氧基或氨基取代;或具有羟基、氨基或卤素基团。在实施方案中,修饰的核苷酸包括甲基胞苷和/或假尿苷。核苷酸可以通过磷酸二酯键或通过合成键(即除磷酸二酯键之外的键)连接。可用于本公开的多核苷酸剂中的核苷间键的实例包括但不限于磷酸二酯、烷基膦酸酯、硫代磷酸酯、二硫代磷酸酯、磷酸酯、烷基硫代膦酸酯、氨基磷酸酯、氨基甲酸酯、碳酸酯、吗啉代、磷酸三酯、乙酰胺酸酯、羧甲基酯或它们的组合。在本公开的实例中,使用了以下rnai剂:
51.非特异性乱序对照sirna:rcrgrururararurcrgrcrgrurarurararurarcrgrcrgruat(seq id no:1)
52.pepd sirna#1:rgrcrarurururgrarurcrargrarcrcrarararcrargrurgct(seq id no:2)
53.pepd sirna#2:rgrgrcrcrgrurcrurarurgrargrgrcrargrurgrcrurgrcgg(seq id no:3)
54.pepd sirna#3:rcrgrarargrurcrararcrararurarcrcrarururcrururcac(seq id no:4)
55.在一个实施方案中,rnai剂包含rgmcra mumumu rgramumcrarg ramcmcrararamcrarg murgc t/36-fam(seq id no:5),在有义链的所有c和u残基处具有2'-o-甲基化。这种或任何其它rnai剂可以具有进一步的修饰以例如增强递送。例如,rnai剂可以与一种或多种蛋白质复合。在实施方案中,rnai剂不可逆地或可逆地附着于蛋白质。在实施方案中,蛋白质包含癌细胞特异性结合配偶体,例如蛋白质或肽,包括但不一定限于癌细胞受体配体,从而可以将rnai剂特异性递送至包含突变pepd的癌细胞。在实施方案中,rnai剂用于与特异性结合以下的配体复合:her2/neu(人表皮生长因子受体2型)、成纤维细胞生长因子受体(fgfr)、e-钙粘蛋白、ema(上皮膜抗原)、αvβ6整合素、epcam(上皮细胞粘附分子)、cea(癌胚抗原)、fr-α(叶酸受体-α)或upar(尿激酶型纤溶酶原激活物受体)、αvβ3整合素、铃蟾肽r、癌胚抗原(cea)、cd13、cd44、c-x-c趋化因子受体-4(cxcr)、碳酸酐酶-9(caix)、细胞外基质金属蛋白酶诱导因子(emmprin)(cd147)、内皮糖蛋白(cd105)、上皮细胞粘附分子(epcam)、met、ifg1r、epha2、成纤维细胞活化蛋白-α(fap-α)、蛋白裂解酶(matriptase)、间皮素、mt1-mmp、muc-1、前列腺干细胞抗原(psca)、前列腺特异性膜抗原(psma)、tn抗原、尿激酶型纤溶酶原激活物受体(upar)或vegfr。在一个实施方案中,rnai剂以与erbb1或erbb2或其片段的复合物提供。在实施方案中,蛋白质或肽配体存在于融合蛋白中。在实施方案中,蛋白质或肽癌细胞特异性配体以包含免疫球蛋白或免疫球蛋白(例如抗体)的片段的融合蛋白提供。这样的蛋白质的非限制性实例包括单链可变片段(scfvs)、vhh单链抗体、fab、单域抗体(sdab、vhh)、亲和体或darpins。在一个实施方案中,配体和免疫球蛋白的片段由接头分开,例如柔性gs接头,其许多合适的实例是本领域已知的。在一个实
施方案中,rnai剂与erbb2和scfv-鱼精蛋白片段(例如氨基酸8-29)的融合蛋白一起使用。在一个实施方案中,鱼精蛋白片段包含氨基酸序列rsqsrsryyrqrqrsrrrrrrs(seq id no:6)或由其组成。在实施方案中,本公开包括包含scrv和精氨酸聚合物(例如,九聚体精氨酸肽)的融合物,例如lu等人,biomaterials 2016,76,196-207中描述的那些,其描述通过引用并入本文。这样的方法和组合物在本领域中是已知的并且可以适于与考虑本公开的本文提供的rnai剂一起使用。(参见,例如,song等人,antibody mediated in vivo delivery of small interfering rnas via cell-surface receptors.nature biotechnology 2005,23,709-717;yao et al.,targeted delivery of plk1-sirnaby scfv suppresses her2 breast cancer growth and metastasis.science translational medicine 2012,4,130ra48;lu等人,sirna delivered by egfr-specific scfv sensitizes egfr-tki-resistant human long cancer cells.biomaterials 2016,76,196-207;其公开内容通过引用并入)。
56.在实施方案中,本公开包括基于患有表达如本文所述的突变p53的癌症来选择癌症患者,和向个体施用靶向pepd的rnai剂,从而破坏pepd和突变p53蛋白之间的相互作用,并为个体提供针对癌症的预防或治疗效果。
57.在实施方案中,癌症患者患有表达如本文所述的突变p53蛋白的任何类型的癌症。因此,癌症的类型没有特别限制,可以包括但不一定限于乳腺癌、前列腺癌、胰腺癌、肺癌、肝癌、卵巢癌、宫颈癌、结肠癌、食道癌、胃癌、膀胱癌、脑癌、睾丸癌、头颈癌、黑色素瘤、皮肤癌、任何肉瘤,包括但不一定限于纤维肉瘤、血管肉瘤、腺癌和横纹肌肉瘤,以及任何血癌,包括所有类型的白血病、淋巴瘤或骨髓瘤。在实施方案中,癌症包括三阴性乳腺癌(tnbc)。tnbc代表缺乏雌激素受体(er)、孕激素受体和her2受体酪氨酸激酶的乳腺癌亚组。目前尚无靶向治疗,患者预后较差。
58.在实施方案中,可以将rnai剂作为裸多核苷酸、与递送剂组合或作为包含和/或表达rnai剂的重组质粒或病毒载体施用于个体。在一个实施方案中,蛋白质由重组溶瘤病毒编码,该病毒可以特异性靶向癌细胞,并且对于非癌细胞可能是非感染性的,和/或如果溶瘤病毒进入非癌细胞则从非癌细胞中消除。
59.在实施方案中,使用治疗上可接受量的rnai剂。治疗有效量是可以达到预期效果的量,例如降低肿瘤生长速率、抑制肿瘤形成、抑制转移或预防癌症和/或肿瘤的发展。
60.在实施方案中,如果需要,本公开的rnai剂可以与递送剂组合。除上述融合蛋白外,用于施用的合适递送试剂包括但不限于mirus transit tko亲脂性试剂;lipofectin;lipofectamine;cellfectin;或聚阳离子(例如聚赖氨酸)、脂质体、纳米颗粒或其组合。在实施方案中,可以通过肿瘤内注射施用rnai剂。在实施方案中,可以使用纳米颗粒或其它合适的药物递送剂,使得rnai剂包含在纳米颗粒中。在实施方案中,纳米颗粒或其它药物递送剂可以与特异性结合癌细胞标志物的结合配偶体一起提供。如上所述,在实施方案中,结合配偶体包括癌细胞表面受体配体。在实施方案中,结合配偶体包括抗体或其抗原结合片段,其可以作为与癌细胞表面受体配体的融合蛋白提供。在实施方案中,当递送使得rnai剂和药物递送剂特异性靶向癌细胞时,施用可以包括任何合适的途径,包括口服、肠胃外、皮下、腹膜内、肺内和鼻内以及颅内。肠胃外输注包括肌肉内、静脉内、动脉内、腹膜内和皮下施用。还包括直接注射到肿瘤中。
61.如本文所述的癌症治疗或抑制可以与任何其它抗癌方法组合,例如手术干预和常规化学治疗剂。在实施方案中,根据本公开的癌症治疗可以与一种或多种免疫检查点抑制剂的施用组合。在实施方案中,检查点抑制剂包含抗程序性细胞死亡蛋白1(抗-pd-1)检查点抑制剂,或抗细胞毒性t淋巴细胞相关蛋白4(抗-ctla-4)检查点抑制剂。本领域已知有许多这样的检查点抑制剂。例如,抗pd-1剂包括派姆单抗(pembrolizumab)和纳武单抗(nivolumab)。一种抗pd-l1的实例是阿维鲁单抗(avelumab)。一种抗ctla-4的实例是伊匹单抗(ipilimumab)。在实施方案中,组合rnai剂和化学治疗剂,或组合rnai剂和免疫检查点抑制剂,可以表现出协同抗癌作用。
62.以下实施例旨在说明本公开的某些实施方案,但并非旨在限制。
63.以下实验使用几种人乳腺癌细胞系进行,包括1)mcf-7(wt-53);2)cal-51(wt-p53);3)bt-474(p53
e285a
);4)mda-mb-231(p53
r280k
);5)具有p53敲除(p53-/-)的mda-mb-231,其通过crispr-cas9从mda-mb-231(p53
r280k
)产生;6)mda-mb-468(p53
r273h
);7)hcc70(p53
r248q
);8)skbr3(p53
r175h
)。mcf-7是雌激素受体阳性乳腺癌细胞系。bt-474和skbr3是her2阳性乳腺癌细胞系。其它细胞系是三阴性乳腺癌细胞系。bt-474细胞中的p53突变体对温度敏感,在37℃显示突变活性,但在32℃恢复为野生型活性。所有实验均在37℃下进行。
64.实施例1
65.如图1所示,该实施例提供了bt-474细胞中的一个热点p53突变体(即p53
e285a
)的结果。通过sirna的pepd kd与p53水平无变化但bcl-xl和bcl-2的下调以及bax和bak的诱导相关(a)。bcl-2家族蛋白质的变化与细胞生长的显著抑制一致(b)。这些结果表明,pepd kd可重新激活p53
e285a
。
66.实施例2
67.该实施例表明,通过sirna的pepd kd可诱导p53
e285a
突变体的转录非依赖性肿瘤抑制活性。特别地,图2中提供的数据显示,通过sirna的pepd kd诱导p53突变体从胞质溶胶和核的线粒体易位(a),线粒体p53突变体与亲环蛋白d(cypd;线粒体通透性转换孔的关键调节因子)的结合(b)和线粒体损伤(线粒体膜电位损失)(c)。
68.实施例3
69.该实施例表明bt-474细胞中p53
e285a
的重新激活。为了确定pepd kd对p53
e285a
转录功能的影响,将bt-474细胞用等量的p53报告子——包含多个拷贝的p53结合序列的pg13-luc或包含多个拷贝的突变p53结合序列的mg15-luc以及用于控制转染效率的prl-tk(renilla luc)转染;24小时后,细胞用对照sirna或pepd sirna(10nm)处理48小时。pg13-luc通过将荧光素酶(luc)表达增加5.9倍来响应pepd ko,而mg15-luc的luc表达在pepd kd后几乎没有变化(图3a)。这表明pepd kd可能显著增加p53
e285a
的反式激活活性,这与如前所述pepd kd对各种p53靶蛋白的调节一致。此外pepd kd刺激p53磷酸化,如通过phostag wb分析检测的(图3b)。测量了p53反式激活结构域中的两个特定磷酸化位点(丝氨酸6和丝氨酸15),并且pepd敲低导致两个位点的磷酸化(图3c)。通过分别分析核级分和胞质级分,我们发现pepd kd诱导的p53磷酸化发生在核p53而不是胞质p53(图3d)。
70.实施例4
71.该实施例证明了bt-474细胞中pepd与p53突变体的结合。我们分析了bt-474细胞中p53
e285a
与pepd的结合。为了估计bt-474细胞中与p53
e285a
结合的细胞pepd的百分比,将全
细胞裂解物中的所有p53
e285a
分子用过量的p53抗体沉淀下来,并且通过比较pepd条带的强度与输入对照条带的强度来确定与p53
e285a
一起下来的pepd分子的百分比(图4a)。我们发现在这些细胞中有约6%的细胞pepd分子与p53
e285a
结合(图4b)。为了估计与pepd结合的细胞p53
e285a
分子的百分比,将细胞裂解物中的所有pepd分子用过量的pepd抗体沉淀下来,并通过比较p53
e285a
条带的强度与输入对照条带的强度来确定保留在上清液中的p53
e285a
分子的百分比(图4c)。我们发现细胞系中约55%的p53
e285a
分子与pepd结合(图4d)。我们还测量了保留在上清液中的p53
e285a
,并且发现该级分中有约45%的突变分子(图4c)。
72.为了更好地了解pepd如何抑制核p53
e285a
,我们用对照或pepd sirna处理bt-474细胞,然后分析核p53
e285a
。pepd敲低导致核p53水平显著降低(图4e)。接下来,通过pepd沉淀去除核提取物中与pepd结合的p53
e285a
。pepd kd不引起与pepd保持结合的p53
e285a
(沉淀部分)的磷酸化(图4e)。然而,尽管p53
e285a
由于pepd kd从核中退出,但在这些细胞中,无pepd的核p53
e285a
水平(上清液部分)显著增加(图4e)。此外,无pepd的核p53
e285a
在pepd kd后磷酸化增加(图4e)。因此,一旦从pepd中释放出来,p53
e285a
就会被磷酸化。
73.实施例5
74.该实施例证明通过crisp-cas9的pepd ko以p53依赖性方式杀死人癌细胞。结果如图5所示,并且表明pepd ko杀死癌细胞需要p53突变体。crispr-cas9核酸酶和grna靶基因敲除组购自celltechgen(ctg-cs9o-19761),包括crispr cas9核酸酶表达载体(pst1374-n-nls-flag-cas9-egfp)、pepd grna载体1(pgl3-pepd-sgrna1)、pepd grna载体2(pgl3-pepd-sgrna2)和乱序rna载体。用于通过crispr-cas9进行pepd敲除的人pepd特异性grna序列作为dna等效物如下:gccgctcacacaggcgctgc(grna1;seq id no:7)和gcggaagaaccctgctgtgc(grna2;seq id no:8)。使用了mda-mb-231(纯合p53
r280k
)及其p53-无效同基因对应物(mda-mb-231p53-/-)。它们是三阴性乳腺癌(tnbc)细胞。mda-mb-231-p53-/-是使用crispr-cas9从mda-mb-231-p53
r280k
产生的(mukhopadhyay等人,j natl cancer inst 2019,111,djz051)。用各种质粒(每个细胞系的质粒量相同)转染细胞并在72小时后检查。
75.实施例6
76.该实施例证明了pepd与p53
wt
和各种p53突变体的结合。特别地,该实施例证明了pepd以相似的亲和力直接与p53突变体结合,并且pepd与每种p53突变体的近一半的胞质和核内含物结合。
77.为了获得这些结果,我们在细菌中产生了带his标签的重组人蛋白,包括pepd、p53及其突变体,通过亲和色谱纯化它们,并通过凝胶电泳和银染证实它们的纯度(见图13a)。我们通过elisa测量了pepd与p53及其突变体的结合。pepd以几乎相同的亲和力与p53
wt
、p53
r175h
、p53
r248q
、p53
r273h
和p53
r280k
结合,显示kd为145-185nm(参见图6a)。我们之前表明,pepd不与p53
mprd
结合,其中prd中除了一个脯氨酸以外的所有脯氨酸都变为丙氨酸(yang等人,nature communications 2017,8,2052)。我们使用p53
mprd
作为阴性对照来确认elisa特异性检测pepd与p53中prd的结合。
78.我们接下来测量了细胞中pepd与p53突变体的结合。我们筛选了7个细胞系,包括skbr3(纯合p53
r175h
)、hcc70(纯合p53
r248q
)、mda-mb-468(纯合p53
r273h
)、mda-mb-231(纯合p53
r280k
)、mda-mb-231(p53
ko
)、mcf-7(p53
wt
)、和cal-51(p53
wt
)。它们是人乳腺癌细胞,her2
阳性(skbr3)、雌激素受体阳性(mcf-7)或三阴性(所有剩余的细胞系)。mda-mb-231(p53
ko
)是通过tp53的crispr-cas9敲除从mda-mb-231产生的。mda-mb-231(p53
r280k
)和mda-mb-231(p53
ko
)构成了同基因的细胞对。我们通过sanger测序确认了每个细胞系中的p53基因型(图13b)。我们通过蛋白质印迹(wb)测量了每个细胞系中p53和pepd二者的表达。如预期的,与p53
wt
相比,所有p53突变体都过表达,并且p53在mda-mb-231-p53
ko
中不存在,但有趣的是,pepd在携带p53突变体的细胞中甚至在mda-mb-231-p53
ko
中也过表达(图6b)。识别p53
wt
及其突变体二者的抗体do-1用于在wb中检测p53。
79.基于这些结果,我们测量了skbr3、mda-mb-231、mda-mb-468和hcc70的全细胞裂解物中pepd与p53突变体的结合,并包括cal-51以与p53
wt
进行比较。由于pepd存在于胞质溶胶和核二者中,我们还测量了pepd与mda-mb-231、skbr3和cal-51的细胞溶质级分和核级分中的p53的结合。用过量抗体对每个样品中的pepd或p53进行免疫沉淀(ib)。通过wb分析上清液证实了pepd或p53的完全沉淀下来(图14a、14b)。通过wb分析沉淀物的p53或pepd以及输入(图14a、14b),并且通过比较它们的条带强度与输入的条带强度来计算相互共沉淀的pepd或p53的百分比。通过对核纤层蛋白b(一种核蛋白)和α-微管蛋白(一种细胞溶质蛋白)的wb分析,排除了胞质溶胶和核提取物之间的交叉污染(图14c)。对于全细胞裂解物,在ip后,我们还通过elisa测量上清液中的pepd或p53,以确定不相互结合的pepd或p53分子的百分比。在沉淀物和相应的上清液中检测到的pepd或p53的组合量占细胞中pepd的95-97%和p53的93-96%(图6c、6d),这验证了我们的实验方法。在全细胞裂解物中有约23%的p53
wt
和40-47%的突变体与pepd结合,在胞质溶胶和核提取物中的结果相似(图6c)。相比之下,只有6-9%的pepd与p53
wt
和突变体结合(图6d),即使pepd的细胞水平是p53细胞水平的3.1-8.7倍高,如通过elisa测量的(图6e)。
80.实施例7
81.该实施例证明了pepd kd对细胞存活和关键蛋白水平的影响,以及pepd
g278d
的反作用。结果如图7所示。r175h、r248q、r273h和r280k是最常见的癌症相关p53突变体,并且是周知的功能获得(gof)突变体。使用了七种细胞系,包括hcc70(纯合p53
r248q
)、mda-mb-468(纯合p53
r273h
)、skbr3(纯合p53
r175h
)、mda-mb-231(纯合p53
r280k
)及其p53-无效同基因对应物(mda-mb-231-p53-/-)、cal-51(p53
wt
)和mcf-7(p53
wt
)。所有细胞系均来自atcc,除了mda-mb-231-p53-/-,它是使用crispr-cas9从mda-mb-231-p53
r280k
产生的(mukhopadhyay等人,j natl cancer inst 2019,111,djz051)。我们用对照或pepd sirna(origene)处理细胞48-72小时。使用分别靶向外显子12(sirna2)和15(sirna1)的两种pepd sirna排除非特异性影响。每种pepd sirna都导致细胞死亡,在72小时处理后杀死64-89%的携带p53
wt
或突变体的细胞(图7a)。然而,两种sirna在mda-mb-231-p53-/-细胞中都没有细胞毒性(图7a)。在所有携带p53突变体或p53
wt
的细胞系中,我们检测到pepd kd后p53靶基因对的强烈调节,包括p21、cd95、puma、bax和bak的诱导,以及bcl-2和bcl-xl的下调,尽管一些bcl-2家族蛋白的调节在细胞系中不统一,但这些变化都没有发生在p53-无效细胞中(图7b)。两种pepd sirna的效果没有差异(图7a、7b、15a)。因此,在后来的实验中,我们专注于pepd sirna1。由于p21在g1/s期抑制细胞周期,我们还通过流式细胞术分析了mda-mb-231、mda-mb-231(p53
ko
)、skrb3和cal-51中的细胞周期。pepd sirna1对mda-mb-231(p53
ko
)中的细胞周期没有影响,但在其它细胞系中引起s期停滞(图15b)。除mda-mb-231(p53
ko
)外,pepd sirna还在
所有细胞系中引起多种胱天蛋白酶(胱天蛋白酶-9、-8、-7)的激活(图7b、15a)。这些结果表明,pepd kd不仅激活p53
wt
,而且重新激活致癌p53突变体。
82.有趣的是,pepd kd仅在p53
wt
细胞中激活胱天蛋白酶3,但在任何携带p53突变体的细胞系中均不激活(图15c)。此外,pepd kd没有改变p53
wt
和突变体的总细胞水平(图7b、15a)。然而,通过使用ip从全细胞裂解物中去除所有pepd,包括与p53结合的pepd,并通过wb测量的上清液的p53,我们发现pepd kd将p53
wt
和突变体从pepd中释放出来(图7c)。
83.我们接下来研究了pepd
g278d
,一种与p53中的prd结合的酶失活pepd突变体(yang等人,nature communications 2017,8,2052),是否可以中和pepd sirna对p53突变体的影响。用表达pepd
g278d
的质粒转染细胞,然后用sirna处理72小时。pepd sirna在没有pepd
g278d
的情况下杀死了65-79%的细胞,但在有pepd
g278d
的情况下仅杀死了16-25%的细胞(图7d)。pepd
g278d
拯救非常有效,因为基因转染效率不太可能达到100%。在表达pepd
g278d
的细胞中,pepd和pepd
g278d
的总水平没有降低,并且尽管具有pepd sirna处理,但p53
wt
靶蛋白(p21和puma)的诱导在很大程度上被取消(图7e)。这些结果进一步证明pepd sirna对p53突变体的重新激活是由于它们与pepd的分离,并表明pepd的酶活性不参与调节p53突变体。
84.实施例8
85.该实施例在同基因细胞中证明了pepd kd对p53突变体的影响。值得注意的是,在细胞中超过50%的p53突变体分子不与pepd结合(图6c)。c-myc(myc)、表皮生长因子受体(egfr)和丝裂原活化蛋白激酶3(mkk3)是由各种p53突变体上调的癌基因。wb分析显示,四种p53突变体中的每一种转染到mda-mb-231(p53
ko
)细胞中都诱导了myc,其中三种还诱导了egfr和mkk3,但没有突变体诱导受p53
wt
调节的cd95和puma(图8a)。然而,如果mda-mb-231(p53
ko
)细胞用p53突变体转染并在24小时后用pepd sirna处理,则台盼蓝测定法显示表达每种突变体的细胞存活被显著抑制(图8b)。wb分析显示pepd sirna没有引起p53水平的变化,造成显著的pepd kd,引起p21、cd95和puma的诱导,并且引起胱天蛋白酶7的激活,但没有引起mkk3、egfr和myc的诱导或者在一些情况下甚至造成下调(图8c)。这些结果与图7中的结果一起表明,pepd kd对p53突变体的重新激活不依赖于细胞环境,并且重新激活的突变分子支配细胞命运,尽管未重新激活的突变分子可能仍发挥促癌活性。
86.实施例9
87.该实施例证明了pepd kd对p53突变体的转录非依赖性肿瘤抑制活性的影响。结果如图9所示,并且与图15和16相关。结果表明,p53(wt或突变体)的线粒体富集伴随着截短的bh3相互作用结构域死亡激动剂(tbid)的线粒体富集,细胞色素c、细胞凋亡诱导因子(aif)和核酸内切酶g(endog)的线粒体损失,细胞色素c和tbid的细胞溶质增加,bid的细胞溶质减少以及aif和endog的核增加(图9a)。已知bid被胱天蛋白酶8转化为促凋亡tbid,其迁移到线粒体。aif和endog的核易位和细胞色素c从线粒体的细胞溶质易位是线粒体介导凋亡的成熟机制。然而,尽管pepd kd在mda-mb-231(p53
ko
)细胞的胞质溶胶和核中均显著,但aif、endog、细胞色素c、bid和tbid没有亚细胞重新分布(图9a)。
88.与上述分子变化一致,jc-1荧光染色表明通过sirna的pepd kd在携带p53
wt
或突变体的细胞中导致线粒体膜电位(mmp)的显著损失,并且mmp损失的程度在不同p53基因型中相似,但pepd kd对mda-mb-231(p53
ko
)中的mmp没有显著影响(图9b)。众所周知,线粒体p53
wt
通过与亲环蛋白d(cypd)结合以打开线粒体通透性转换孔并通过与bcl-2和bcl-xl结
合以中和它们对bak和bax的抑制作用,从而导致mmp损失和细胞死亡。通过co-ip和wb,我们发现pepd sirna显著增加了所有p53突变体与线粒体中cypd的结合(图9c)。然而,由于pepd kd导致bcl-2、bcl-xl、bak和bax的表达发生显著变化(图7b),因此评估p53突变体与线粒体中bcl-2家族蛋白的相互作用是不可行的。我们还在选择的细胞系中进行了tunel测定法,结果表明pepd sirna在携带p53
wt
或突变体的细胞中强烈诱导细胞凋亡,但在p53
ko
细胞中无活性(图9d、15d)。
89.已知的是,p53
wt
易位至线粒体是由mdm2介导的单泛素化驱动的。还已知低活性水平的mdm2会导致p53
wt
的单泛素化,但高活性水平的mdm2导致p53
wt
的多泛素化和降解。专注于skbr3细胞中的p53
r175h
,我们发现通过sirna的mdm2 kd阻断了pepd kd诱导的p53
r175h
线粒体富集(图16a),增加了mdm2与p53
r175h
的结合(图16b),并在线粒体中富集了单泛素化p53
r175h
(图16c)。然而,pepd kd不诱导核p53
r175h
的单泛素化(图16c)。
90.实施例10
91.该实施例证明了pepd kd对p53突变体的磷酸化和转录活性的影响。结果如图10和图17所示。pepd kd显然也重新激活了p53突变体的转录功能(图7b)。因为磷酸化是激活p53
wt
转录活性的一个众所周知的关键步骤,我们通过wb测量了p53
wt
和突变体的反式激活结构域中的四个磷酸化位点(s6、s15、s20、s46)。pepd kd诱导了p53
wt
和所有突变体的磷酸化,但磷酸化位点在它们之间有所不同(图10a、17a)。变异性至少部分地与细胞环境有关,因为p53
wt
中的磷酸化位点在mcf-7和cal-51之间也不同(图10a、17a)。因为没有共有的磷酸化位点,我们接下来使用检测整体磷酸化的phostag wb来确定磷酸化-p53的细胞位置。我们专注于p53
r280k
(mda-mb-231)、p53
r175h
(skbr3)和p53
wt
(cal-51)。pepd kd诱导的磷酸化-p53(wt和突变体)发生在核中,但不在胞质溶胶或线粒体中(图10b)。我们通过ip从核提取物中去除了所有pepd,并测量了沉淀物和上清液中的磷酸化p53,但仅在上清液中检测到了磷酸化-p53(wt和突变体)(图17b),表明p53在离开pepd后被磷酸化。接下来,用pg13-luc或mg15-luc以及用于控制转染效率的prl-tk转染细胞,然后用sirna处理48小时。pepd sirna在cal-51细胞(p53
wt
)中诱导来自pg13-luc的报告子表达4.2倍,在携带p53突变体的细胞中诱导3.3-5.6倍(图10c)。相比之下,pepd sirna在所有细胞中对来自mg15-luc的报告子表达的影响可以忽略不计。使用chip-qpcr,我们进一步表明,在用pepd sirna处理48小时的细胞中,每种p53突变体与编码p21的cdkn1a基因启动子中的p53
wt
结合位点的结合增加了7.6-10.2倍,并且与编码puma的bbc3基因的启动子中的p53
wt
结合位点的结合增加了5.9-7.6倍(图10d)。
92.总的来说,上述结果以及之前显示的其它结果表明,p53突变体一旦被pepd kd从pepd中释放出来,就会被磷酸化并通过与其启动子中的p53
wt
结合位点结合来调节p53
wt
靶基因。p53突变体重新激活的转录活性与激活的p53
wt
几乎没有区别。
93.实施例11
94.该实施例显示了pepd kd对p53突变体的再折叠和重新激活的影响,以及k373乙酰化的作用。结果显示在图11和图18中。结果表明k373的乙酰化对于p53突变体的重新激活是必不可少的,并且p53突变体一旦与pepd分离就重新折叠,这是由k373乙酰化驱动的。众所周知,p53
wt
的乙酰化对其激活至关重要。我们通过wb测量了pepd kd对p53突变体各种赖氨酸残基乙酰化的影响。通过sirna的pepd kd在不同位点诱导乙酰化,但仅k373乙酰化是所
有突变体共有的,没有k370乙酰化,并且k372乙酰化变化很小(图11a)。专注于skbr3细胞(p53
r175h
)和mda-mb-231细胞(p53
r280k
),我们发现k373乙酰化发生在核和线粒体中而不是胞质溶胶中的突变体(图11b、18a)。我们接下来询问k373乙酰化是发生在突变体离开pepd之前还是之后。因为线粒体中不存在pepd,我们分析了核提取物。我们使用ip从核提取物中去除所有pepd,并通过wb测量沉淀物和上清液中的ac-k373-p53突变体。由pepd kd产生的ac-k373-p53突变体仅存在于上清液中(图18b)。因此,k373乙酰化发生在突变体离开pepd之后。已知c646抑制使p53乙酰化的p300/cbp。c464阻止pepd sirna引起携带p53突变体的所有细胞系死亡(图11c、18c)。c464还在所有细胞系中阻断了p53突变体的k373乙酰化和p53
wt
靶蛋白(包括p21和puma)的诱导,尽管有强烈pepd kd(图11d、18d)。为了进一步阐明k373乙酰化在p53突变体重新激活中的作用,我们将k373r突变引入每种p53突变体并在mda-mb-231(p53
ko
)细胞中表达突变体,然后通过sirna进行pepd kd 96小时。k373r突变使每种p53突变体几乎完全不响应pepd kd,如通过细胞存活以及p21和puma的诱导所评估的(图11e、11f、18e、18f)。因此,k373乙酰化是pepd kd重新激活p53突变体的关键分子开关。
95.我们想知道pepd是否与p53突变体结合以将它们支架化回野生型构象。抗体pab1620和pab240分别广泛用于检测p53的“野生型”和“变性”构象,尽管它们不能告知蛋白质的精确折叠状态。我们确认pab1620和pab240分别专门检测“野生型”和“变性”构象(图18g)。已知p53
r175h
完全以“变性”构象存在。事实上,ip接着wb表明,skbr3细胞中的p53
r175h
不与pab1620结合,而是与pab240结合(图11g、11h)。pepd与p53
r175h
一起被pab240沉淀下来(图11h),表明p53
r175h
在与pepd结合后仍处于“变性”构象。在pepd sirna处理48小时后,约40%的p53
r175h
分子从“变性”构象转变为“野生型”构象(图11i)。约40%p53
r175h
的重新折叠与skbr3中最初与pepd结合的p53
r175h
的百分比非常匹配(图6c),表明所有p53
r175h
分子在离开pepd后重新折叠。我们还检查了p53
r248q
、p53
r273h
和p53
r280k
。通过pab240或pab1620对全细胞裂解物进行ip,通过wb分析上清液证实了每种p53突变体的完全沉淀(图18h)。通过wb对沉淀物的分析表明,每种突变体主要以“野生型”构象存在,其余为“变性”构象(图18h),这与之前的报道一致。pepd与每种p53突变体一起被pab1620和pab240二者沉淀,但主要是pab1620(图18h)。因此,无论其构象如何,pepd都与p53突变体结合,并且不会诱导突变体重新折叠。然而,pepd kd增加了三种突变体的“野生型”构象水平(图18i),并且相对较小的增加与最初以“变性”构象存在的相对较小的突变分子池非常匹配。值得注意的是,pepd kd不会改变p53突变体的总水平(图7b)。因此,处于“变性”构象的突变分子在离开pepd后可能都变为“野生型”构象。总的来说,我们的结果表明,pepd与p53突变体的结合不会改变它们的构象,但突变体在离开pepd后会重新折叠
96.c646阻断了pepd kd诱导的所有四种p53突变体从“变性”到“野生型”的构象变化(图11j、18j)。我们接下来在mda-mb-231(p53
ko
)细胞中表达p53
r175h/k373r
、p53
r248q/k373r
、p53
r273h/k373r
或p53
r280k/k373r
,然后用乱序或pepd sirna处理该细胞。k373r突变没有改变每种p53突变体与pepd的结合,但完全阻断了由pepd kd诱导的突变体的构象变化(图11k、18k)。因此,在p53突变体离开pepd后发生的k373乙酰化驱动它们的重新折叠。此外,使用p53
r175h/k373r
和p53
r280k/k373r
,我们发现,当在mda-mb-231(p53
ko
)细胞中表达时,在用pepd sirna处理细胞时两种双突变体均未在线粒体中富集(图11l、18l)。总的来说,k373乙酰化是pepd kd重新激活p53突变体的转录依赖性和非依赖性肿瘤抑制功能的基础。
97.实施例12
98.该实施例显示了在有或没有p53突变体表达的同基因肿瘤中pepd kd对生长和关键蛋白的表达的体内影响。结果显示在图12中,并且与图19相关。数据表明,在具有由mda-mb-231-p53
r280k
、mda-mb-231-p53
ko
或mda-mb-231-p53
r175h
细胞产生的原位乳腺肿瘤的相关动物模型中,通过肿瘤内注射pepd sirna进行的pepd kd诱导的重新激活的p53突变体强烈抑制肿瘤生长。
99.我们首先比较了pepd kd对仅p53不同的同基因原位肿瘤的影响。我们将mda-mb-231细胞(p53
r280k
)或mda-mb-231(p53
ko
)细胞接种到雌性scid小鼠的乳腺脂肪垫上。一旦肿瘤达到约100mm3,就每三天一次给予瘤内注射乱序或pepd sirna (10pmol)。当对照小鼠的平均肿瘤尺寸达到约550mm3时停止实验,以尽量减少sirna分布到肿瘤组织中的障碍。分别在最终治疗后1天和2天从小鼠收集mda-mb-231(p53
ko
)肿瘤和mda-mb-231(p53
r280k
)肿瘤。我们没有检测到sirna对小鼠的不利影响。在乱序sirna下两种类型的肿瘤都迅速生长。然而,pepd sirna强烈抑制mda-mb-231(p53
r280k
)肿瘤的生长,并且在实验结束时,平均肿瘤尺寸和肿瘤重量分别仅为对照组的10.6%和8.6%(图12a、12b)。相比之下,pepd sirna对mda-mb-231(p53
ko
)肿瘤的生长没有影响(图12c、12d)。通过wb对代表性肿瘤样品的分析显示,两种类型的肿瘤之间的pepd水平相似,并且pepd sirna在两种肿瘤中引起了明显的pepd kd。在mda-mb-231(p53
r280k
)肿瘤中,pepd sirna没有改变总p53水平,但强烈上调p21、cd95和bak,下调bcl-2并活化胱天蛋白酶7(图12e),这与体外检测到的变化非常相似(图7b)。相比之下,在mda-md-231(p53
ko
)肿瘤中,pepd sirna对p21、cd95、bcl-2、bak和胱天蛋白酶7没有影响(图12e)。
100.因为p53
r280k
是接触突变体,我们还研究了p53
r175h
,它是一种构象突变体并且完全“变性”。skrb3细胞携带p53
r175h
但不能在体内产生肿瘤,无论小鼠品系(scid、nod scid或裸鼠)或接种途径(皮下或原位)如何。由于在mda-mb-231(p53
ko
)细胞中瞬时表达的p53
r175h
被pepd kd重新激活(图8b、8c),我们使用这些细胞来产生稳定表达p53
r175h
的mda-mb-231细胞。通过wb筛选了多种p53
r175h
表达克隆,并选择了p53
r175
水平与skbr3的相似的克隆#1(图19a)。mda-mb-231(p53
r175h
)的优势在于它与mda-mb-231(p53
ko
)同基因,后者在体外和体内对pepd kd均无响应。我们首先证实了pepd sirna在体外强烈抑制mda-mb-231(p53
r175h
)细胞的生长(图19b)。我们接下来将mda-mb-231(p53
r175h
)细胞接种到雌性scid小鼠的乳腺脂肪垫上,并且肿瘤迅速生长。当肿瘤达到约130mm3时,开始瘤内注射乱序或pepd sirna(10pmol),每三天给予一次。一旦对照小鼠的平均肿瘤尺寸达到约550mm3,就停止实验。在最终治疗后两天从小鼠中收集肿瘤。同样,sirna对小鼠没有不利影响。尽管在乱序sirna中肿瘤生长迅速,但pepd sirna强烈抑制肿瘤生长,并且在实验结束时,平均肿瘤尺寸和肿瘤重量分别仅为对照组的28.9%和25.9%(图12f、12g)。通过wb对代表性肿瘤样品的分析表明,pepd sirna引起明显的pepd kd,不改变p53水平,但强烈上调p21、cd95和bak,下调bcl-2并激活胱天蛋白酶7(图12e),与p53
r175h
的重新激活一致。
101.值得注意的是,在来源于人结肠癌hct116细胞(p53
wt
)和hct116细胞(p53
ko
)的小鼠的一对同基因肿瘤中,通过肿瘤内注射sirna的pepd kd抑制p53
wt
肿瘤的生长达79%,同时强烈激活p53靶基因但对p53
ko
肿瘤没有影响(yang等人,nature communications 2017,8,2052)。因此,pepd kd重新激活的p53
r175h
和p53
r280k
的体内肿瘤抑制活性与pepd kd激活
的p53
wt
的体内肿瘤抑制活性相似。
102.实施例13
103.该实施例提供了pepd、pepd突变体、p53
wt
、p53突变体和细胞系的表征。结果如图13所示,并且与图6和图7相关。
104.实施例14
105.该实施例证明了细胞中pepd与p53
wt
及其突变体的结合。结果如图14所示,并且与图6相关。
106.实施例15
107.该实施例证明了pepd kd对p53和其它蛋白质水平、细胞周期进程和细胞凋亡的影响。结果如图15所示,并且与图7和图9相关。
108.实施例16
109.该实施例证明了mdm2在skbr3细胞中p53
r175h
的线粒体富集中的作用。结果如图16所示,并且与图9相关。
110.实施例17
111.该实施例证明了pepd kd对p53
wt
和p53突变体的磷酸化的影响。结果如图17所示,并且与图10相关。
112.实施例18
113.该实施例证明了pepd kd对p53突变体的重新折叠的影响,以及k373乙酰化在p53突变体的重新折叠和重新激活中的作用。结果如图18所示,并且与图11相关。
114.实施例19
115.该实施例提供了稳定表达p53
r175h
的mda-mb-231细胞的特征。结果如图19所示,并且与图12相关。
116.上述实施例旨在说明某些实施方案,但不限制本公开的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。