一种rass-f1甲基化检测组合物及其应用
技术领域
1.本发明属于生物技术领域,涉及一种rass-f1甲基化检测组合物及其应用。
背景技术:
2.肺癌的五年生存率仅15%,早发现对患者的治疗极为重要。目前,对肺癌的诊断主要依据临床症状、医学影像学检测和组织病理学检查等,但是许多临床症状在中晚期才出现,同时活体取样检测也十分困难,严重影响了肺癌的早期诊断和治疗预后。
3.随着生物技术的发展,肿瘤标志物检测日益成为辅助肿瘤诊断、监测、用药指导和治疗评估的有效手段。肺癌在早期阶段由于无特殊症状,难以用常规方法进行检测,而一些肿瘤标志物仅能辅助诊断提示,无法用于早期筛查。dna甲基化检测给肺癌的早期诊断与筛查带来了希望。dna甲基化几乎存在于所有肿瘤中,成为肿瘤诊断的一个可靠靶标。dna甲基化往往发生在早期阶段,在疾病确诊之前就能被检测出来,是肿瘤早期诊断、风险评估的一个重要指标,甲基化作为重要的肿瘤标志物,优势在于:肿瘤形成过程中甲基化发生频率很高,通常发生于肿瘤的早期阶段,且甲基化稳定存在于样本中,便于检测。
4.rass-f1是dammann等于2000年在肺癌和乳腺癌细胞株中克隆的一个新型候选抑癌基因,是ras相关区域家族1基因,因其表达产物中含有与ras蛋白结构相关的区域,故而得名。目前,普遍认为rass-f1基因表达失活与启动子区特异性高甲基化、杂合性丢失和染色体缺失有关,尤其是rassfla基因启动子甲基化与肿瘤的关系是目前研究的热点。
5.在人类肿瘤中,rassfla基因失活的频率很高。例如,rassfla在70%以上小细胞肺癌、91%肾细胞癌、62%膀胱癌、7l%甲状腺癌、84%鼻咽癌和70%以上前列腺癌等中处于甲基化状态进而失活。现已明确rassfla基因在肺癌中表达失活的机制主要是由于其启动子区cpg岛的特异性高甲基化。虽然抑癌基因的失活还可能由突变所致,但是对该基因的研究结果表明,rassfla基因的突变率在肺癌中还不及10%,而rassfla启动子区发生高甲基化的比例在小细胞肺癌、非小细胞肺癌中分别高达100%和63%。
6.目前,主要利用重亚硫酸盐转化法对rass-f1基因甲基化进行检测。重亚硫酸盐转化法的原理是采用重亚硫酸盐处理dna,将胞嘧啶残基转化成尿嘧啶,而被甲基化的胞嘧啶残基不受影响,因此,重亚硫酸盐处理后的dna片段只保留有甲基化胞嘧啶。基于此原理,重亚硫酸盐能在单个核苷酸水平上揭示dna的甲基化情况。已有多种检测方法可以实现对重亚硫酸盐处理后dna序列的分析,分析的实际问题就是重亚硫酸盐使碱基从c到u最终到t造成的区别。
7.但是,重亚硫酸盐转化法也存在诸多不足:(1)重亚硫酸盐测序需要确保重亚硫酸盐转化反应完全,即每一个未被甲基化的胞嘧啶都被转化为尿嘧啶,如果转化反应不完全则会出现假阳性结果;(2)由于只有单链dna的胞嘧啶才能被重亚硫酸盐攻击,在转化前需要对dna进行变性解链,必须严格控制温度、盐浓度等因素,否则会造成转化失败或转化不完全;(3)dna在转化反应过程中有可能被降解,如果孵育时间过长、温度和重亚硫酸盐浓度过高,可能导致高达90%的dna被降解,降解后的dna脱嘌呤形成随机断裂,可能导致pcr扩
增失败或dna样本数量少,准确性低,进而出现检测假阴性;(4)重亚硫酸盐处理会显著降低样本的复杂性,使得多重pcr引物设计更加困难,误差增加。
8.因此,发展一种高灵敏度dna甲基化检测方法,不需重亚硫酸盐处理、不需设置对照反应,成为迫切需要解决的问题。
技术实现要素:
9.针对现有技术的不足和实际需求,本发明提供了一种rass-f1甲基化检测组合物及其应用,基于甲基化依赖型限制性内切酶和通用引物,并采用荧光定量pcr技术,检测rass-f1基因特殊位点上的甲基化情况,具有不需要重亚硫酸盐转化、操作简单、准确性高和特异性强的优点。
10.为达此目的,本发明采用以下技术方案:
11.第一方面,本发明提供了一种rass-f1甲基化检测组合物,所述组合物包括甲基化依赖型限制性内切酶、捕获寡核苷酸和通用引物;
12.所述捕获寡核苷酸从5’端到3’端依次包括第一通用序列、折叠序列和结合捕获序列;
13.所述折叠序列与rass-f1甲基化位点经甲基化依赖型限制性内切酶酶切后的5’末端序列至少部分相同;
14.所述结合捕获序列与所检测的rass-f1甲基化位点所在的片段区域特异性结合。
15.优选的,所述捕获寡核苷酸还包括第二通用序列。
16.优选地,所述第二通用序列位于结合捕获序列的5’端。
17.本发明中,rass-f1甲基化检测组合物主要包括甲基化依赖型限制性内切酶和基于通用引物的荧光定量pcr检测试剂两部分:甲基化依赖型限制性内切酶通过对rass-f1甲基化模板的甲基化位点进行酶切处理,形成5’端序列明确的中间产物;捕获寡核苷酸利用结合捕获序列捕获该中间产物,并以此作为模板进行延伸反应,在捕获寡核苷酸的3’末端添加与rass-f1甲基化基因互补的核苷酸,从而形成延伸的捕获寡核苷酸,延伸的捕获寡核苷酸通过延伸序列与其自身内部的折叠序列进行完全匹配或非完全匹配的配对,形成半发夹结构产物,半发夹结构产物再进行延伸反应,在3’末端添加与其自身内部的第一通用序列互补的核苷酸,进而形成完整的发夹结构产物;所述发夹式结构产物利用通用引物和/或特异性引物进行pcr扩增,结合检测探针实现了rass-f1甲基化位点的荧光定量pcr检测。
18.优选地,所述捕获寡核苷酸还包括核酸延伸阻断位点。
19.优选地,所述核酸延伸阻断位点位于折叠序列的3’端,用于隔断结合捕获序列和折叠序列。
20.优选地,所述核酸延伸阻断位点修饰有spacer、硫代基团或尿嘧啶碱基中的任意一种或至少两种的组合。
21.优选地,所述折叠序列修饰有核酸类似物。
22.优选地,所述核酸类似物包括肽核酸、锁核酸、转置碱基、2'-o,4'-c-methylene bridge rna、2
’‑
o-methyl rna或2
’‑
fluoro rna中的任意一种或至少两种的组合。
23.优选地,所述通用引物的核酸序列与捕获寡核苷酸的第一通用序列相同或部分相同。
24.优选地,所述甲基化依赖型限制性内切酶包括glai、fspei、mspji、lpnpi、aspbhi或msei中的任意一种或至少两种的组合。
25.优选地,所述组合物还包括rass-f1甲基化特异性引物。
26.优选地,所述组合物还包括检测探针。
27.优选地,所述检测探针标记有荧光基团和/或淬灭基团。
28.优选地,所述荧光基团标记在检测探针的5’端。
29.优选地,所述淬灭基团标记在检测探针的3’端。
30.优选地,所述荧光基团包括fam、vic、joe、tet、cy3、cy5、rox、texas red或lc red460中的任意一种。
31.优选地,所述淬灭基团包括bhq1、bhq2、bhq3、dabcy1或tamra中的任意一种。
32.优选地,所述捕获寡核苷酸包括seq id no:1、seq id no:6或seq id no:8所示的核酸序列。
33.优选地,所述通用引物包括seq id no:2或seq id no:9所示的核酸序列。
34.优选地,所述rass-f1甲基化特异性引物包括seq id no:3或seq id no:7所示的核酸序列。
35.优选地,所述检测探针包括seq id no:4所示的核酸序列。
36.第二方面,本发明提供了一种rass-f1甲基化检测试剂盒,所述试剂盒包括第一方面所述的组合物。
37.优选地,所述试剂盒还包括dna聚合酶、dntps或mg
2
中的任意一种或至少两种的组合。
38.优选地,所述试剂盒还包括酶切缓冲液和/或pcr缓冲液。
39.第三方面,本发明提供了一种rass-f1甲基化检测体系,所述体系包括1~20nm捕获寡核苷酸、100~400nm通用引物、100~300nm检测探针、1~2u taq聚合酶、100~300μm dntp、1~5mm mgcl2和pcr缓冲液。
40.优选地,所述体系还包括100~300nm rass-f1甲基化特异性引物。
41.第四方面,本发明提供了一种rass-f1甲基化检测方法,所述方法包括:
42.采用甲基化依赖型限制性内切酶对待测rass-f1甲基化模板进行酶切处理,将酶切处理后的产物加入第三方面所述的体系中,进行荧光定量pcr。
43.优选地,所述酶切处理的温度为30~40℃,例如可以是30℃、31℃、32℃、33℃、34℃、35℃、36℃、37℃、38℃、39℃或40℃,优选为37℃。
44.优选地,所述酶切处理的时间为0.5~2h,例如可以是0.5h、1h、1.5h或2h,优选为1h。
45.优选地,所述荧光定量pcr的程序为92~95℃预变性2~5min;92~95℃变性10~20s,65~70℃退火80~100s,10~15个循环;92~95℃变性10~20s,65~70℃退火20~30s,30~50个循环。
46.第五方面,本发明提供了一种rass-f1甲基化检测装置,所述装置包括:
47.酶切处理单元:采用甲基化依赖型限制性内切酶对待测rass-f1甲基化模板进行酶切处理,获得5’端序列明确的预处理产物;
48.荧光定量pcr单元:利用含有捕获寡核苷酸、通用引物、rass-f1甲基化特异性引物
和检测探针的荧光定量pcr体系对预处理产物进行荧光定量pcr检测。
49.优选地,所述酶切处理单元提供的酶切处理温度为30~40℃,例如可以是30℃、31℃、32℃、33℃、34℃、35℃、36℃、37℃、38℃、39℃或40℃,优选为37℃。
50.优选地,所述酶切处理单元提供的酶切处理时间为0.5~2h,例如可以是0.5h、1h、1.5h或2h,优选为1h。
51.优选地,所述荧光定量pcr单元提供的荧光定量pcr程序为92~95℃预变性2~5min;92~95℃变性10~20s,65~70℃退火80~100s,10~15个循环;92~95℃变性10~20s,65~70℃退火20~30s,30~50个循环。
52.第六方面,本发明提供了第一方面所述的组合物、第二方面所述的试剂盒、第三方面所述的体系或第五方面所述的装置在制备疾病早期诊断试剂和/或设备中的应用。
53.优选地,所述疾病包括肿瘤。
54.优选地,所述肿瘤包括肺癌、肝癌、肾细胞癌、膀胱癌、甲状腺癌、鼻咽癌、前列腺癌或结直肠癌中的任意一种或至少两种的组合。
55.与现有技术相比,本发明具有如下有益效果:
56.(1)本发明的rass-f1甲基化检测组合物主要包括甲基化依赖型限制性内切酶和基于通用引物的荧光定量pcr检测试剂两部分,不需要对甲基化样本进行连接反应、化学处理等额外步骤,仅需在pcr检测之前对样本进行预处理,得到5’端序列明确的样本,结合基于通用引物的扩增技术,实现了核酸的高特异、高灵敏的多重检测;
57.(2)本发明的rass-f1甲基化检测组合物中,捕获寡核苷酸与通用引物经过特殊设计,相互配合,在靶标分子存在的情况下,靶标分子触发由捕获寡核苷酸介导的延伸反应,形成发夹式结构产物,并以此作为通用引物扩增反应的模板,由于扩增反应基于5’序列确定的产物,有效避免了假阳性问题;
58.(3)本发明中,当捕获寡核苷酸的3’延伸序列与折叠序列能够形成互补配对,才能引发捕获寡核苷酸的自我折叠形成发夹式结构,有效保证了反应的特异性;
59.(4)本发明的rass-f1甲基化检测试剂盒在针对不同靶标分子进行检测时,只需要根据靶标分子设计捕获寡核苷酸的折叠序列和结合捕获序列,而保持第一通用序列不变,极大程度降低多重靶标扩增的多种引物间干扰,提高扩增的灵敏度;
60.(5)本发明的rass-f1甲基化检测试剂盒通过指数扩增过程达到信号放大的目的,不仅能很好地满足dna/rna检测时对灵敏度的需求,而且所述指数扩增过程仅利用延长的捕获寡核苷酸和通用引物来完成,可在保持通用引物的数量和浓度不变的前提下达到对多重靶标分子的等效扩增目的,避免了序列差异导致的扩增效率的偏差;
61.(6)本发明的rass-f1甲基化检测方法操作简便。
附图说明
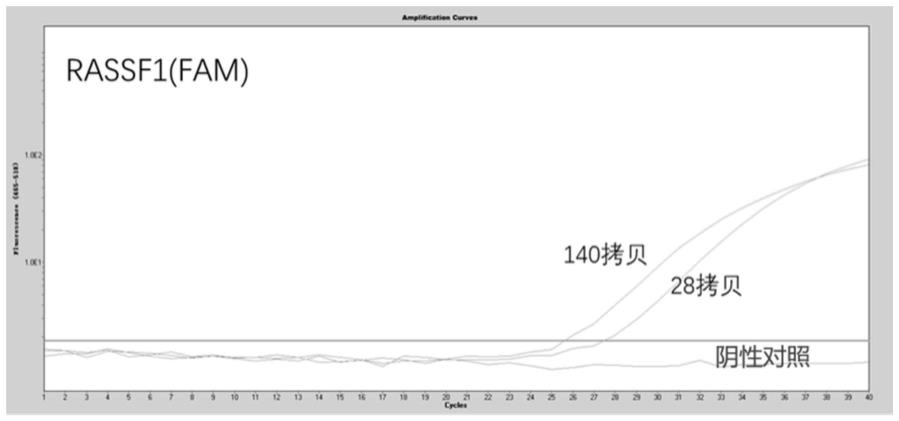
62.图1为不同浓度的甲基化样本中rass-f1基因的灵敏度检测结果;
63.图2为不同浓度的甲基化阳性样本rass-f1基因的甲基化状态;
64.图3为rass-f1基于双通用引物的扩增结果。
具体实施方式
65.为进一步阐述本发明所采取的技术手段及其效果,以下结合实施例和附图对本发明作进一步地说明。可以理解的是,此处所描述的具体实施方式仅仅用于解释本发明,而非对本发明的限定。
66.实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。
67.实施例1人源rass-f1基因的甲基化dna检测
68.本实施例采用甲基化转移酶处理的jurkat dna作为甲基化阳性标准品,无核酸酶水作为阴性对照,其中cg位点为5mcg,进行rass-f1基因的甲基化检测,甲基化依赖型限制性内切酶采用gla i。步骤如下:
69.配制含有2μl 10
×
酶切缓冲液、5u gla i、不同浓度基因组dna(或阴性对照)的20μl反应体系,37℃孵育1小时进行酶切反应,得到5’端序列明确的预处理产物;酶切反应结束后,将体系加热至85℃孵育10min,对glai进行热灭活;
70.分别向上述酶切反应体系中加入rass-f1基因捕获寡核苷酸、通用引物、特异性引物和检测探针,pcr检测rass-f1基因的甲基化状态,采取的pcr扩增体系包括酶切dna模板、10nm捕获寡核苷酸、200nm通用引物、200nm特异性引物、200nm检测探针、1u taq聚合酶、200μm dntp、4.5mm mgcl2和2
×
pcr缓冲液,终体积为20μl;pcr反应程序为94℃预变性5min;94℃变性10s,66℃退火90s,10个循环;94℃变性10s,65℃退火20s,40个循环;在roche仪器(480)上进行实时pcr,采用相应的荧光数值;
71.捕获寡核苷酸(seq id no:1):
72.5-tgtcagccaacggtattcatcgcccagcgggtgcgaagcacgggcccaac;
73.通用引物(seq id no:2):
74.5-tgtcagccaacggtattcatc;
75.特异性引物(seq id no:3):
76.5-ccatgtcgggggagcctg;
77.检测探针(seq id no:4):
78.5-fam-ctcccgcagctcaatg-mgb;
79.人源rass-f1基因如seq id no:5所示,其中,下划线为甲基化位置,//为酶切位置:
80.5-gcgc//gcccagcgggtgccagctcccgcagctcaatgagctcaggctcccccgacatggcccggttgggcccgtgcttcgctgg。
81.如图1所示为不同浓度的甲基化样本中rass-f1基因的灵敏度检测,从左至右分别为140拷贝/反应和28拷贝/反应的dna甲基化阳性样本,通过qpcr曲线可以看出,低至20拷贝左右的dna甲基化样本能被酶切扩增检测到,说明该体系具有较高的灵敏度。
82.实施例2rass-f1折叠区域序列不完全相同的扩增结果
83.本实施例对人源rass-f1基因的甲基化dna进行检测,步骤如下:
84.(1)利用甲基化转移酶处理jurkat dna,构建完全甲基化的阳性基因组,测序鉴定rass-f1基因的甲基化状态;
85.(2)采用甲基化依赖型限制性内切酶glai分别对甲基化阳性jurkat基因组dna(或阴性对照)进行酶切反应,反应体系为10
×
酶切缓冲液2μl,5u glai,不同浓度的基因组dna,体系共20μl;反应条件为37℃孵育1小时;酶切反应结束后,将体系加热至85℃,孵育10分钟,对glai进行热灭活;
86.(3)分别向上述酶切反应体系中加入rass-f1基因捕获寡核苷酸、通用引物、特异性引物和检测探针,pcr检测rass-f1基因的甲基化状态,采取的pcr扩增体系包括酶切dna模板、10nm捕获寡核苷酸、200nm通用引物、200nm特异性引物、200nm检测探针、1u taq聚合酶、200μm dntp、4.5mm mgcl2和2
×
pcr缓冲液,终体积为20μl;pcr反应程序为94℃预变性5min;94℃变性10s,66℃退火90s,10个循环;94℃变性10s,65℃退火20s,40个循环;在roche仪器(480)上进行实时pcr,对相应的荧光数值进行采集;
87.捕获寡核苷酸(seq id no:6):
88.5-tgtcagccaacggtattcatcgcccag c ggtttttgccag/spacer 18/gcgaagcacgggcccaac( 后面的碱基代表该碱基修饰锁核酸);
89.通用引物(seq id no:2):
90.5-tgtcagccaacggtattcatc;
91.特异性引物(seq id no:7):
92.5-ccatgtcgggggagcctgag;
93.检测探针(seq id no:4):
94.5-fam-ctcccgcagctcaatg-mgb;
95.人源rass-f1基因如seq id no:5所示。
96.本实施例中,捕获寡核苷酸的折叠区域与靶标分子5’末端序列不完全相同。采用qpcr检测不同浓度的甲基化阳性样本rass-f1基因的甲基化状态,如图2所示,曲线1、2、3为rass-f1甲基化样本的扩增曲线,分别为120拷贝/反应、40拷贝/反应及12拷贝/反应的rass-f1甲基化基因样本,阴性对照为添加ddh2o的扩增曲线。
97.由此说明,本发明的捕获寡核苷酸折叠区域与靶标分子5’末端序列只需有部分序列相同,即可实现扩增。
98.实施例3rass-f1基于双通用引物的扩增结果
99.本实施例对人源rass-f1基因的甲基化dna进行检测,步骤如下:
100.(1)利用甲基化转移酶处理jurkat dna,构建完全甲基化的阳性基因组,测序鉴定rass-f1基因的甲基化状态;
101.(2)采用甲基化依赖型限制性内切酶glai分别对甲基化阳性jurkat基因组dna(或阴性对照)进行酶切反应,反应体系为10
×
酶切缓冲液2μl,5u gla i,不同浓度的基因组dna,体系共20μl;反应条件为37℃孵育1小时;酶切反应结束后,将体系加热至85℃,孵育10分钟,对glai进行热灭活;
102.(3)分别向上述酶切反应体系中加入rass-f1基因捕获寡核苷酸、通用引物、特异性引物和检测探针,pcr检测rass-f1基因的甲基化状态,采取的pcr扩增体系包括酶切dna模板、10nm捕获寡核苷酸、200nm第一通用引物、200nm第二通用引物、200nm检测探针、1u taq聚合酶、200μm dntp、4.5mm mgcl2和2
×
pcr缓冲液,终体积为20μl;pcr反应程序为94℃预变性5min;94℃变性10s,66℃退火90s,10个循环;94℃变性10s,65℃退火20s,40个循环;
在roche仪器(480)上进行实时pcr,对相应的荧光数值进行采集;
103.捕获寡核苷酸(seq id no:8):
104.tgtcagccaacggtattcatcgcccagcgggtgcca/spacer18/agatgtcgcactgacaacgggccatgtcgggggag;
105.第一通用引物(seq id no:2):
106.tgtcagccaacggtattcatc;
107.第二通用引物(seq id no:9):
108.cggcgtcagatgtcgcactgacaa;
109.检测探针(seq id no:4):
110.5-fam-ctcccgcagctcaatg-mgb。
111.本实施例中,去除了来自于模板的特异性引物序列,在捕获寡核苷酸的结合区域和折叠区域之间再加入一段通用引物序列。采用qpcr检测不同浓度的甲基化阳性样本rass-f1基因的甲基化状态,如图3所示,曲线1、2、3、4为rass-f1甲基化样本的扩增曲线,分别为1200拷贝/反应、120拷贝/反应、40拷贝/反应及12拷贝/反应的rass-f1甲基化基因样本,阴性对照为添加ddh2o的扩增曲线。从扩增曲线看出,低至40拷贝/反应的阳性模板能进行扩增,12拷贝/反应有微弱扩增。
112.由此说明,本发明使用双通用引物体系也能有效的实现低浓度rass-f1甲基化基因的检测。
113.综上所述,本发明采用甲基化依赖型限制性内切酶和基于通用引物的pcr扩增相结合的方式,不需重亚硫酸盐处理、不需设置对照反应,实现了rass-f1甲基化dna的精确定量检测,避免了序列差异导致的扩增效率的偏差,特异性好,适于推广应用。
114.申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
