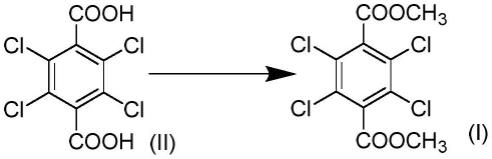
1.本发明涉及2,3,5,6-四氯-1,4-苯二甲酸二甲酯的制造方法。
背景技术:
2.如下述反应式1所示,2,3,5,6-四氯-1,4-苯二甲酸二甲酯(下述式(i))通过以下方法制造:将2,3,5,6-四氯-1,4-苯二甲酸(下述式(ii))用甲醇和三氧化硫/硫酸/氯磺酸进行甲酯化的方法(专利文献1)、将2,3,5,6-四氯-1,4-苯二甲酸在氢氧化钾水溶液中用硫酸二甲酯进行单甲酯化后将未反应的羧基用甲醇/强酸进一步酯化的方法(专利文献2)、或者在非水系且丙酮中将2,3,5,6-四氯-1,4-苯二甲酸在硫酸二甲酯和碳酸钠的存在下进行甲酯化的方法(非专利文献1)。
3.[化1]
[0004]
(反应式1)
[0005][0006]
另外,如下述反应式2所示,2,3,5,6-四氯-1,4-苯二甲酸二甲酯(下述式(i))可以通过将2,3,5,6-四氯-1,4-苯二甲酰氯(下述式(iv))用甲醇和碱进行甲酯化的方法(专利文献3)来制造。
[0007]
[化2]
[0008]
(反应式2)
[0009][0010]
在上述反应式1所示的2,3,5,6-四氯-1,4-苯二甲酸二甲酯的制造方法中,使用由对苯二甲酸的环上氯代而得到的2,3,5,6-四氯-1,4-苯二甲酸作为原料。原料2,3,5,6-四氯-1,4-苯二甲酸的制造方法如以往技术的专利文献4~6以及非专利文献2和3所示。
[0011]
另外,在专利文献7中,作为上述2,3,5,6-四氯-1,4-苯二甲酸的其他制造方法,记载了如下方法:将2,3,5,6-四氯-1,4-苯二甲腈的cn基转化为酰胺基而得到2,3,5,6-四氯-1,4-苯二甲酰胺,然后使用发烟硫酸等制造2,3,5,6-四氯对苯二甲酸。
[0012]
另一方面,在反应式2所示的2,3,5,6-四氯-1,4-苯二甲酸二甲酯的制造方法中,以2,3,5,6-四氯对苯二甲酰二氯为原料,该原料通过将对苯二甲酰氯进行环上氯代而得到(专利文献8~13,非专利文献4和5)。
[0013]
现有技术文献
[0014]
专利文献
[0015]
专利文献1:us3689526a
[0016]
专利文献2:us3689527a
[0017]
专利文献3:特开昭60-16952
[0018]
专利文献4:us3873613a
[0019]
专利文献5:su352882a1
[0020]
专利文献6:de1078563b
[0021]
专利文献7:us2001/0025121a1
[0022]
专利文献8:特开昭48-013339
[0023]
专利文献9:特开昭51-138641
[0024]
专利文献10:特开昭58-157727
[0025]
专利文献11:us3052712a
[0026]
专利文献12:us3833652a
[0027]
专利文献13:us4808345a
[0028]
非专利文献:
[0029]
非专利文献1:zhurnal prikladnoi khimii,(1978),51(6),1422-1423
[0030]
非专利文献2:vop.neftekhim,(1971),no.3,49-51
[0031]
非专利文献3:zhurnal obshchei khimii,(1964),34(9),2953-2958
[0032]
非专利文献4:gaofenzi xuebao,(2004),(5),773-775
[0033]
非专利文献5:yingyong huaxue,(2005),22(4),317-390
技术实现要素:
[0034]
发明要解决的课题
[0035]
关于上述反应式1的制造方法,专利文献1和2所述的制造方法均是复杂的,因为是2阶段的反应。另外,在非专利文献1所述的制造方法中,存在未反应的原料残留,反应未完结的问题。
[0036]
采用上述专利文献4~6以及非专利文献2和3所述的2,3,5,6-四氯-1,4-苯二甲酸的制造方法时,大量生成了不理想的副产物六氯苯,得到的2,3,5,6-四氯-1,4-苯二甲酸中含有大量的六氯苯。当使用由此得到的含有大量六氯苯的2,3,5,6-四氯-1,4-苯二甲酸作为原料来制造2,3,5,6-四氯-1,4-苯二甲酸二甲酯时,在得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯中六氯苯以不可接受的浓度存在。
[0037]
上述专利文献7所述的2,3,5,6-四氯-1,4-苯二甲酸的制造方法虽然可抑制六氯苯等副产物的大量生成,但却是采用两阶段反应的制造方法,不仅复杂,而且由于使用发烟硫酸等,导致原料的处理伴随着风险。
[0038]
在上述专利文献8~13以及非专利文献4和5所述的对苯二甲酰氯的环上氯代中,难以控制六氯苯的生成,得到的2,3,5,6-四氯对苯二甲酰二氯中含有大量的六氯苯。当使用含有大量六氯苯的2,3,5,6-四氯对苯二甲酰二氯来制造2,3,5,6-四氯-1,4-苯二甲酸二甲酯时,得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯中,六氯苯以不可接受的浓度存在。
[0039]
本发明的目的在于,在制造用作农业园艺用除草剂的2,3,5,6-四氯-1,4-苯二甲
酸二甲酯时,提供与以往方法相比降低了六氯苯、五氯苯等对环境有害的副产物的含量、能够有效制造的工业化方法。
[0040]
解决课题的手段
[0041]
本发明人等为了解决上述课题,进行了反复深入研究,结果发现,通过将洗涤2,3,5,6-四氯-1,4-苯二甲酸二甲酯的晶体的条件设定为特定条件,实现了大幅减少副产物六氯苯和五氯苯的含量的效果,完成了本发明。
[0042]
本发明的具体实施方式如下所示。
[0043]
[1]式(i)表示的化合物的制造方法,
[0044]
[化3]
[0045][0046]
其包括以下工序:
[0047]
(a)将式(ii)表示的化合物在含水酮系溶剂中在碱金属碳酸盐的存在下与硫酸二甲酯反应,由此得到作为晶体的式(i)表示的化合物的工序,
[0048]
[化4]
[0049][0050]
和
[0051]
(b)将上述晶体用30~100℃的温水洗涤,然后再用30~80℃的有机溶剂洗涤的工序。
[0052]
[2]上述[1]所述的方法,其中,上述工序(b)是将上述晶体在加温下用有机溶剂洗涤,由此减少上述晶体中含有的有害副产物的含量的工序。
[0053]
[3]上述[1]或[2]所述的方法,其中,在上述工序(b)中,在用温水洗涤后且用有机溶剂洗涤之前的上述晶体的温度为40~90℃。
[0054]
[4]上述[1]~[3]任一项所述的方法,其中,上述温水的温度为60~95℃。
[0055]
[5]上述[1]~[4]任一项所述的方法,其中,上述工序(b)的上述有机溶剂为醇。
[0056]
[6]上述[1]~[5]任一项所述的方法,其中,上述工序(b)的上述有机溶剂为甲醇、乙醇、异丙醇、或它们的混合物。
[0057]
[7]上述[1]~[6]任一项所述的方法,其中,上述工序(b)的上述有机溶剂为甲醇。
[0058]
[8]上述[5]~[7]任一项所述的方法,其中,上述有机溶剂的温度为35~65℃。
[0059]
[9]上述[1]~[8]任一项所述的方法,其在上述工序(a)之前,还包括(a’)将式
(iii)表示的化合物在酸的存在下加热至100~180℃而得到上述式(ii)表示的化合物的工序,
[0060]
[化5]
[0061][0062]
[10]上述[9]所述的方法,其中,将上述工序(a’)得到的上述式(ii)表示的化合物用水洗涤后,在上述工序(a)中使用。
[0063]
[11]上述[1]~[10]任一项所述的方法,其中,上述工序(a)的上述含水酮系溶剂为含水丙酮。
[0064]
[12]上述[1]~[11]任一项所述的方法,其中,上述工序(a)的上述含水酮系溶剂的含水量为5~25重量%。
[0065]
[13]上述[1]~[12]任一项所述的方法,其中,上述工序(a)的上述碱金属碳酸盐为碳酸钠。
[0066]
[14]上述[2]所述的方法,其中,上述有害的副产物为多氯苯类。
[0067]
[15]上述[14]所述的方法,其中,上述多氯苯类为六氯苯、五氯苯、或它们的混合物。
[0068]
[16]一种组合物,其包含式(i)表示的化合物,
[0069]
[化6]
[0070][0071]
其所含有的六氯苯的含量大于0ppm且为40ppm以下,其所含有的五氯苯的含量大于0ppm且为1000ppm以下。
[0072]
[17]上述[16]所述的组合物,其用作除草剂或除草剂原料。
[0073]
[18]上述[16]或[17]所述的组合物,其中,上述式(i)表示的化合物为晶体状态。
[0074]
[19]上述[16]~[18]任一项所述的组合物,其中,上述式(i)表示的化合物相对于上述组合物全体的含量为96.0~100重量%。
[0075]
发明效果
[0076]
根据本发明,在制造用作农业园艺用除草剂的2,3,5,6-四氯-1,4-苯二甲酸二甲酯时,能够有效地制造与以往方法相比六氯苯和五氯苯等有害副产物的含量少的目标物。
具体实施方式
[0077]
以下,详细说明本发明的式(i)表示的化合物的制造方法。其中,本发明不限定于
以下记载。
[0078]
本发明是上述式(i)表示的化合物的制造方法,其包括以下工序:(a)将上述式(ii)表示的化合物在含水酮系溶剂中在碱金属碳酸盐的存在下与硫酸二甲酯反应,由此得到作为晶体的式(i)表示的化合物的工序,和(b)将上述晶体用30~100℃的温水洗涤,然后再用30~80℃的有机溶剂洗涤的工序。
[0079]
(工序(a))
[0080]
工序(a)中使用的式(ii)表示的化合物(2,3,5,6-四氯-1,4-苯二甲酸)可通过公知的制造方法制造,例如可通过后述的工序(a’)制造。
[0081]
工序(a)中使用的含水酮系溶剂没有特殊限定,优选为含水丙酮、含水2-丁酮、含水3-戊酮、或它们的混合物,其中,从与水的亲和性和成本的观点考虑,特别优选为含水丙酮。
[0082]
含水酮系溶剂的含水量没有特殊限定,优选为5~25重量%,更优选为10~20重量%,最优选为15~20重量%。本发明中含水酮系溶剂的含水量是指使用卡尔费歇尔-电滴定装置,基于jis k0068所述的条件,通过容量滴定法测定的值。当含水酮系溶剂的含水量在上述数值范围内时,发挥反应完成且目标产物的收量稳定的效果。
[0083]
含水酮系溶剂的含水量可通过向工序(a)的反应混合物中加入水来调制。
[0084]
工序(a)中使用的含水酮系溶剂的添加量相对于式(ii)表示的化合物1kg,优选为1.0~5.0l,更优选为1.0~3.0l,最优选为1.2~2.0l。
[0085]
工序(a)中使用的碱金属碳酸盐没有特殊限定,优选为碳酸钠、碳酸钾、碳酸铯、或它们的混合物,其中,从成本和收率的观点考虑,特别优选为碳酸钠。
[0086]
工序(a)中使用的碱金属碳酸盐的添加量相对于式(ii)表示的化合物,优选为1.0~5.0当量,更优选为1.0~3.0当量,最优选为1.2~2.0当量。
[0087]
工序(a)中使用的硫酸二甲酯的添加量相对于式(ii)表示的化合物,优选为1.5~4.0当量,更优选为1.7~3.0当量,最优选为2.0~2.5当量。
[0088]
工序(a)中的反应温度没有特殊限定,优选溶剂可回流的温度,具体地,优选为40~100℃,更优选为50~80℃,最优选为55~65℃。
[0089]
工序(a)中的反应时间没有特殊限定,优选为2~10小时,更优选为4~8小时,最优选为4~6小时。
[0090]
优选从含有由工序(a)得到的式(i)表示的化合物的晶体的反应混合物中蒸馏除去含水酮系溶剂。蒸馏除去含水酮系溶剂的条件没有特殊限定,关于压力,优选在常压或减压的条件下进行,另外,关于温度,优选加温,可使用温水或蒸气来进行该加温。
[0091]
通过向含有由工序(a)得到的式(i)表示的化合物的晶体的反应混合物中加入40~50℃的温水来冷却,可以使式(i)表示的化合物的晶体析出。由此析出的晶体可通过过滤来回收。
[0092]
由工序(a)得到的式(i)表示的化合物的晶体可通过过滤来回收。过滤的方法没有特殊限定,可使用减压过滤、加压过滤或离心过滤。过滤的条件没有特殊限定,可采用减压或加压的条件。
[0093]
(工序(b))
[0094]
工序(b)中使用的温水的温度为30~100℃,优选为60~95℃,更优选为85~90℃。
当温水的温度在上述数值范围内时,可加温(加热)式(i)表示的化合物的晶体,可将加温状态的晶体进行后续的使用有机溶剂的洗涤处理。
[0095]
工序(b)中使用的温水的量相对于式(ii)表示的化合物1kg,优选为0.5~3.0l,更优选为1.0~2.5l,最优选为1.2~2.0l。
[0096]
工序(b)中,在加入温水之后且在加入有机溶剂之前的式(i)表示的化合物的晶体的温度优选为40~90℃,更优选为50~80℃,最优选为60~75℃。当晶体的温度在上述数值范围内时,可将加温状态的晶体进行后续的使用有机溶剂的处理。
[0097]
工序(b)中使用的有机溶剂的温度为30~80℃,优选为35~65℃,更优选为40~50℃。当有机溶剂的温度在上述数值范围内时,可有效降低式(i)表示的化合物的晶体中含有的有害副产物的含量。
[0098]
工序(b)中使用的有机溶剂没有特殊限定,醇是有效的,优选为甲醇、乙醇、或异丙醇,其中,从成本和洗涤效率的观点考虑,特别优选为甲醇。工序(b)中使用的有机溶剂也可以为上述的甲醇、乙醇和异丙醇中的2种以上的混合物。工序(b)中使用的有机溶剂可使用通用产品,另外,该有机溶剂的纯度优选为100%,也可以为50~100%、70~100%、80~100%、90~99.99%、或95~99.9%。
[0099]
工序(b)中使用的有机溶剂的量相对于式(ii)表示的化合物1kg,优选为0.5~3.0l,更优选为0.7~2.0l,最优选为0.8~1.5l。
[0100]
工序(b)中的使用有机溶剂的洗涤可分1~3次来洗涤,更优选分2次来洗涤。
[0101]
在本发明的一实施方式中,工序(b)优选为通过将式(i)表示的化合物的晶体在加温下用有机溶剂洗涤,以降低上述晶体中含有的有害副产物的含量的工序。
[0102]
在本发明的工序(b)中,通过将式(i)表示的化合物的晶体用30~100℃的温水洗涤来加温晶体,将该加温状态的晶体进一步用30~80℃的有机溶剂洗涤,由此可有效降低式(i)表示的化合物的晶体中含有的有害副产物的含量。
[0103]
关于工序(b),在使用有机溶剂的洗涤后,可以对式(i)表示的化合物的晶体进行离心分离或过滤,其中,优选进行过滤。式(i)表示的化合物的晶体的过滤方法没有特殊限定,优选使用上下真空滤器过滤。作为上下真空滤器过滤,可以使用包括以下工序的过滤:过滤工序、滤饼碾压/压搾工序、滤饼洗涤工序、再浆化洗涤工序、干燥(通气或真空)工序、滤饼排出工序、或这些工序中的1种以上的组合。通过使用上下真空滤器过滤,能够使晶体在被有机溶剂充分浸渍的状态下脱液,因而洗涤效果提高。
[0104]
工序(b)中,在用有机溶剂洗涤后,可对回收的式(i)表示的化合物的晶体进行干燥处理。干燥处理的条件没有特殊限定,关于温度,优选为20~150℃,更优选为40~120℃,最优选为60~100℃,另外,关于压力,优选为2~760mmhg,更优选为10~200mmhg,最优选为20~100mmhg。
[0105]
从式(i)表示的化合物的晶体中除去/减少的有害副产物没有特殊限定,例如为多氯苯类。另外,这样的多氯苯类没有特殊限定,例如为六氯苯、五氯苯、或它们的混合物。
[0106]
经过工序(b)后的式(i)表示的化合物的晶体中含有的六氯苯的含量优选为40ppm以下,更优选为10ppm以下,最优选为5ppm以下。
[0107]
经过工序(b)后的式(i)表示的化合物的晶体中含有的五氯苯的含量优选为1000ppm以下,更优选为500ppm以下,最优选为100ppm以下。
[0108]
本发明中,经过工序(b)后的式(i)表示的化合物的晶体中含有的六氯苯的含量可以在气相色谱装置(产品名称:agilent 7890a,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:hp-5,色谱柱长度30m,色谱柱直径0.53mmid,膜厚1.0μm,安捷伦科技公司制)在fid条件下通过绝对校准曲线法来计算,或者可以在气相色谱-质谱分析装置(产品名称:agilent 7890a gc/5975c msd,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:rxi-5silms,色谱柱长度30m,色谱柱直径0.25mmid,膜厚0.25μm,
レステック
株式会社制)通过绝对校准曲线法来计算m/z283.8的质量数的含量。
[0109]
另外,经过工序(b)后的式(i)表示的化合物的晶体中含有的五氯苯的含量可以在气相色谱装置(产品名称:agilent 7890a,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:hp-5,色谱柱长度30m,色谱柱直径0.53mmid,膜厚1.0μm,安捷伦科技公司制)在fid条件下通过绝对校准曲线法来计算。
[0110]
(工序(a’))
[0111]
本发明的式(i)表示的化合物的制造方法在上述工序(a)之前还可以包括(a’)将上述式(iii)表示的化合物在酸的存在下加热至100~180℃而得到上述式(ii)表示的化合物的工序。
[0112]
工序(a’)中使用的式(iii)表示的化合物(2,3,5,6-四氯-1,4-苯二甲腈)可通过公知的制造方法来制造,例如,工业上可通过使1,4-苯二甲腈与氯反应来制造。
[0113]
工序(a’)中使用的酸没有特殊限定,优选为硫酸、发烟硫酸、氯磺酸、或它们的混合物,其中,从容易操作、容易获得和成本的观点考虑,优选为硫酸,特别优选为98%硫酸。
[0114]
工序(a’)中使用的酸相对于式(iii)表示的化合物的量,按重量比计,优选为2.0~10.0倍,更优选为3.0~8.0倍,最优选为4.0~6.0倍。
[0115]
工序(a’)中的反应温为100~180℃,优选为140~170℃,更优选为155~165℃。
[0116]
工序(a’)中的反应时间没有特殊限定,优选为4~18小时,更优选为5~12小时,最优选为6~9小时。
[0117]
如上所述,专利文献7所述的2,3,5,6-四氯对苯二甲酸的制造方法由于使用发烟硫酸,因此原料的处理伴随着危险。另一方面,在本发明的工序(a’)中,代替发烟硫酸,可采用更简便和温和的条件来进行反应。
[0118]
可以向包含经过工序(a’)后的式(ii)表示的化合物的反应混合物中加入水。通过以此方式加入水,水解反应充分进行,最终能够得到收率和纯度均高的式(i)表示的化合物。添加水的方法没有特殊限定,优选为以硫酸水溶液的形式添加的方法。添加的硫酸水溶液没有特殊限定,从与使用水的情况相比为简便而温和的条件的观点考虑,优选为60~70重量%的硫酸水溶液。
[0119]
通过向包含经过工序(a’)后的式(ii)表示的化合物的反应混合物中加入30~50℃的温水来冷却,能够使式(ii)表示的化合物的晶体析出。由此析出的晶体可通过过滤来回收。
[0120]
对包含经过工序(a’)后的式(ii)表示的化合物的反应混合物进行离心分离处理,由此能够分离得到式(ii)表示的化合物。
[0121]
工序(a’)得到的式(ii)表示的化合物可通过水进行洗涤。用水的洗涤可使用离心分离装置来进行。在用水进行洗涤时,可以进行精制和/或干燥处理,但也可以不进行精制
和/或干燥处理而仅用水进行洗涤。在此,精制可通过使用有机溶剂进行重结晶来实施,另外,干燥例如可使用锥型干燥装置等在加热减压的条件下进行。如上所述,不进行精制和/或干燥处理而仅用水进行洗涤后的式(ii)表示的化合物可原样用于后续的工序(a)中。
[0122]
在经过工序(a’)后的后续工序(a)中使用时的式(ii)表示的化合物的含水量优选为0~15重量%,更优选为1~10重量%,最优选为2~6重量%。
[0123]
(包含式(i)表示的化合物的组合物)
[0124]
本发明的一实施方式为包含式(i)表示的化合物的组合物,该组合物中含有的六氯苯的含量大于0ppm且为40ppm以下,该组合物中含有的五氯苯的含量大于0ppm且为1000ppm以下。
[0125]
包含式(i)表示的化合物的组合物中含有的六氯苯的含量大于0ppm且为40ppm以下,另外,也可以为0.1ppm以上30ppm以下,0.2ppm以上20ppm以下,或0.5ppm以上10ppm以下。
[0126]
包含式(i)表示的化合物的组合物中含有的五氯苯的含量大于0ppm且为1000ppm以下,另外,也可以为1ppm以上500ppm以下,5ppm以上250ppm以下,或10ppm以上150ppm以下。
[0127]
包含式(i)表示的化合物的组合物中含有的六氯苯的含量可以在气相色谱装置(产品名称:agilent 7890a,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:hp-5,色谱柱长度30m,色谱柱直径0.53mmid,膜厚1.0μm,安捷伦科技公司制)在fid条件下通过绝对校准曲线法来计算,或者可以在气相色谱-质谱分析装置(产品名称:agilent 7890a gc/5975c msd,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:rxi-5silms,色谱柱长度30m,色谱柱直径0.25mmid,膜厚0.25μm,
レステック
株式会社制)通过绝对校准曲线法来计算出m/z283.8的质量数的含量。
[0128]
包含式(i)表示的化合物的组合物中含有的五氯苯的含量可以通过在气相色谱装置(产品名称:agilent 7890a,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:hp-5,色谱柱长度30m,色谱柱直径0.53mmid,膜厚1.0μm,安捷伦科技公司制)在fid条件下通过绝对校准曲线法来计算。
[0129]
包含式(i)表示的化合物的组合物的用途没有特殊限定,可用作除草剂或除草剂原料。
[0130]
式(i)表示的化合物的状态没有特殊限定,优选为结晶状态。
[0131]
式(i)表示的化合物相对于组合物全体的含量没有特殊限定,可以为96.0~100重量%,97.0~99.9重量%,或98.0~99.9重量%。
[0132]
实施例
[0133]
以下示出本发明的实施例,但它们仅是用于说明的示例,本发明的范围由权利要求书的范围确定,不受下述记载的任何限制。
[0134]
《实施例1,比较例1~3》
[0135]
[实施例1]从2,3,5,6-四氯-1,4-苯二甲腈(式(iii)表示的化合物)的2,3,5,6-四氯-1,4-苯二甲酸二甲酯(式(i)表示的化合物)的制造
[0136]
(工序1)
[0137]
在具有玻璃衬里的反应器中,将日本燐酸株式会社制的硫酸(98重量%,6348g)加
温至75℃。向反应器中逐渐投入株式会社sds生物技术制的2,3,5,6-四氯-1,4-苯二甲腈(纯度98.5重量%,1380g),期间,将硫酸溶液维持在85~100℃。投入后,将反应混合物在100~120℃下搅拌30分钟。然后,加热至155~163℃,再搅拌4小时。然后,在155~163℃下向反应混合物中滴入硫酸水溶液(62重量%,70g),搅拌2小时,再在155~163℃下滴入硫酸水溶液(62重量%,70g),搅拌1小时。最后在155~163℃下向反应混合物中滴入硫酸水溶液(62重量%,255g),搅拌1小时。然后,将反应混合物冷却至70~80℃,一边将反应混合物的液温维持在低于110℃,一边向其中投入水(3170ml)。投入后,将反应混合物冷却至35~45℃的温度,通过过滤来回收析出的固体。将得到的固体(1611g)用水(2600ml)洗涤。由此得到的固体为2,3,5,6-四氯-1,4-苯二甲酸的晶体(1609g),水含量为4重量%。将该晶体原样用于后续工序2。
[0138]
(工序2)
[0139]
向具有玻璃衬里的反应器中投入丙酮(2033ml)和水(300ml)至丙酮的含水量为15重量%,然后投入工序1中得到的2,3,5,6-四氯-1,4-苯二甲酸(1609g)和东京化成工业株式会社制的碳酸钠(754g)。将反应混合物加热至57℃,一边将反应液温维持在55~58℃的范围,一边滴入东京化成工业株式会社制的硫酸二甲酯(1510g)。然后,一边加热回流反应混合物,一边搅拌4.5小时,然后,在常压下蒸馏除去丙酮(1400ml)。然后,向50~63℃温度下的反应混合物中加入40℃的温水(2550ml),通过过滤来回收析出的固体。将得到的晶体(1777g)用80~90℃的温水(2550ml)洗涤,再用40℃的甲醇(1500ml)洗涤。予以说明,在用温水洗涤之后且在用甲醇洗涤之前的晶体的温度为69℃。将用甲醇洗涤后的晶体通过上下真空滤器过滤来回收,将回收的晶体在80℃、40mmhg的条件下减压干燥,得到2,3,5,6-四氯-1,4-苯二甲酸二甲酯。
[0140]
关于得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯,其收量:1615g,总收率:93.7重量%,纯度:99.6重量%,对环境有害的副产物六氯苯的含量为1.0ppm,五氯苯的含量为40ppm。
[0141]
予以说明,在本实施例中,目标产物的纯度可以在气相色谱装置(产品名称:agilent 7890a,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:hp-5,色谱柱长度30m,色谱柱直径0.53mmid,膜厚1.0μm,安捷伦科技公司制)在fid条件下通过内部标准法来计算。另外,五氯苯的含量使用与上述计算纯度的情况同样的气相色谱装置和毛细管色谱柱在fid条件下通过绝对校准曲线法来计算。六氯苯的含量可以在气相色谱-质谱分析装置(产品名称:agilent 7890a gc/5975c msd,安捷伦科技公司制)中使用毛细管色谱柱(产品名称:rxi-5silms,色谱柱长度30m,色谱柱直径0.25mmid,膜厚0.25μm,
レステック
株式会社制)通过绝对校准曲线法来计算m/z283.8的质量数的含量。通过该测定方法得到的六氯苯的含量和五氯苯的含量的定量限为:六氯苯0.2ppm,五氯苯20ppm。
[0142]
[比较例1]从2,3,5,6-四氯-1,4-苯甲酰胺的2,3,5,6-四氯-1,4-苯二甲酸二甲酯的制造
[0143]
(工序1)
[0144]
本工序1的制造方法是基于上述专利文献7的制造方法。向玻璃反应器中投入株式会社sds生物技术制的2,3,5,6-四氯-1,4-苯二甲酰胺(6.04g,0.02mol),和光纯药株式会社制的硫酸(包含96.3重量%,12.43g,0.0256mol的水)以及和光纯药株式会社制的发烟硫
酸(包含26重量%,5.22g,0.017mol的so3))的混合物(合计:17.65g)。将反应混合物在常压下加热至180℃,搅拌6小时。反应结束后,通过过滤来回收析出的固体,将得到的固体(7.0g)用水(100ml)洗涤。将水洗得到的固体干燥,由此得到2,3,5,6-四氯-1,4-苯二甲酸的白色晶体(5.8g)。将得到的晶体原样用于后续工序2。
[0145]
(工序2)
[0146]
将工序1得到的化合物的白色晶体(5.8g)混悬在甲醇(17ml)中,在室温下用7分钟向反应混合物中滴入氢氧化钠(1.43g)的甲醇(12ml)溶液,然后,一边加热回流2小时一边搅拌。反应结束后,冷却至室温,通过过滤来回收析出的固体。将得到的固体用室温(20℃)的水充分洗涤,然后通过过滤来回收洗涤后的固体,将回收的固体在80℃、40mmhg的条件下减压干燥,得到2,3,5,6-四氯-1,4-苯二甲酸二甲酯(5.2g)。
[0147]
关于得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯,其纯度:99.1重量%,六氯苯的含量为25ppm,五氯苯的含量为500ppm。
[0148]
[比较例2]从2,3,5,6-四氯-1,4-苯二甲腈的2,3,5,6-四氯-1,4-苯二甲酸二甲酯的制造
[0149]
(工序1)
[0150]
本工序(1)的制造方法是基于上述专利文献7的制造方法。向玻璃反应器中投入株式会社sds生物技术制的2,3,5,6-四氯-1,4-苯二甲腈(纯度98.5重量%,5.32g,0.02mol)、和光纯药株式会社制的硫酸(90重量%,11.06g)以及和光纯药株式会社制的发烟硫酸(26重量%,6.59g)的混合物(合计:17.65g)。在常压下加热至160℃,搅拌3小时。反应结束后,通过过滤来回收析出的固体,将得到的固体(7.1g)用水(100ml)洗涤。通过将水洗得到的固体干燥,得到2,3,5,6-四氯-1,4-苯二甲酸的晶体(5.9g)。
[0151]
(工序2)
[0152]
如下所示,将工序1得到的化合物通过非专利文献1所述的方法进行二甲基酯化。
[0153]
将工序1得到的2,3,5,6-四氯-1,4-苯二甲酸的晶体(5.9g)、丙酮(30.3g)、东京化成工业株式会社制的硫酸二甲酯(4.89g)和东京化成工业株式会社制的碳酸钠(2.67g)的混合物在加热回流下搅拌8小时。反应后,将反应混合物冷却至室温,加入水(30g),将析出的固体通过过滤来回收。将得到的固体(7.1g)用室温(20℃)的水100g洗涤。洗涤后的固体为2,3,5,6-四氯-1,4-苯二甲酸二甲酯、2,3,5,6-四氯-1,4-苯二甲酸甲酯、和未反应的2,3,5,6-四氯-1,4-苯二甲酸的混合物(5.9g)。关于得到的混合物,六氯苯的含量为21ppm,五氯苯的含量为400ppm。
[0154]
[比较例3]从2,3,5,6-四氯-1,4-苯二甲腈(式(iii)表示的化合物)的2,3,5,6-四氯-1,4-苯二甲酸二甲酯的制造
[0155]
关于实施例1的工序2的过滤后的晶体的洗涤,代替80~90℃的温水(2550ml)和40℃的甲醇(1500ml),使用室温(20℃)的水(2550ml)和20℃的甲醇(1500ml),除此以外,与实施例1的工序1和工序2同样操作,得到2,3,5,6-四氯-1,4-苯二甲酸二甲酯。予以说明,在用室温(20℃)的水洗涤之后且在用甲醇洗涤之前的晶体的温度为22℃。关于得到的式(i)表示的化合物,六氯苯的含量为10ppm,五氯苯的含量为300ppm。
[0156]
如实施例1所示,在从工业生产的原料2,3,5,6-四氯-1,4-苯二甲腈工业化制造2,3,5,6-四氯-1,4-苯二甲酸的工序(工序1)中,仅使用市售的98重量%的硫酸并且在170℃
以下的反应温度下反应充分进行,能够高收率地生成目标产物。另外,在后续工序的甲酯化反应(工序2)中通过使用含水丙酮,反应也顺利进行,能够以高收率仅得到目标的2,3,5,6-四氯-1,4-苯二甲酸二甲酯。
[0157]
另外,作为实施例1的最终工序(工序2)的目标产物2,3,5,6-四氯-1,4-苯二甲酸二甲酯的洗涤方法,通过用80~90℃的温水洗涤、再用40℃的甲醇洗涤,能够有效地去除对环境有害的六氯苯和五氯苯等。
[0158]
另一方面,在比较例1中,仅用以往采用的室温(20℃)的水来进行最终工序(工序2)的目标产物2,3,5,6-四氯-1,4-苯二甲酸二甲酯的洗涤,已知这样的晶体的洗涤方法不能充分除去对环境有害的六氯苯和五氯苯等。
[0159]
另外,在比较例2中,在工序2的甲酯化反应中使用无水丙酮时,反应在中途停止,得到的是未反应物、单甲酯体和目标产物2,3,5,6-四氯-1,4-苯二甲酸二甲酯的混合物,不能充分得到目标产物2,3,5,6-四氯-1,4-苯二甲酸二甲酯。
[0160]
在仅最终工序(工序2)的晶体洗涤条件与实施例1不同的比较例3中,作为最终工序中的目标产物2,3,5,6-四氯-1,4-苯二甲酸二甲酯的洗涤方法,由于用比实施例1温度低的、室温(20℃)的水和20℃的甲醇洗涤,不能充分除去对环境有害的六氯苯和五氯苯等。
[0161]
因此可知,通过使用市售的98重量%的硫酸将2,3,5,6-四氯-1,4-苯二甲腈转化为2,3,5,6-四氯-1,4-苯二甲酸、在后续工序的甲酯化反应中以含水丙酮为溶剂、以及将目标产物2,3,5,6-四氯-1,4-苯二甲酸二甲酯用30~100℃的温水洗涤,然后再用30~80℃的有机溶剂洗涤,由此,能够工业化制造高纯度的且减少了对环境有害的杂质的含量的目标产物。
[0162]
《实施例2~10、比较例4~8》
[0163]
从2,3,5,6-四氯-1,4-苯二甲腈制造2,3,5,6-四氯-1,4-苯二甲酸二甲酯时的洗涤条件的研究
[0164]
作为原料,使用与实施例1的工序1同样的硫酸和2,3,5,6-四氯-1,4-苯二甲腈,通过与实施例1的工序1同样的方法和条件来制造,分离出含水量为4重量%的2,3,5,6-四氯-1,4-苯二甲酸的晶体(1600g)。将得到的晶体分成每份200g,分别作为批次1~8。将各批次原样用于后续工序。
[0165]
进而,通过与上述同样的方法,制备含水量为4重量%的2,3,5,6-四氯-1,4-苯二甲酸的晶体(1200g)。将得到的晶体分成每份200g,分别作为批次9~14。将各批次原样用于后续工序。
[0166]
[实施例2]
[0167]
向具有玻璃衬里的反应器中投入丙酮(254ml)和水(37ml)至丙酮的含水量为15重量%,然后投入2,3,5,6-四氯-1,4-苯二甲酸(批次1,200g)和东京化成工业株式会社制的碳酸钠(94g),将反应混合物升温至57℃,向55~58℃温度下的反应混合物中滴入东京化成工业株式会社制的硫酸二甲酯(189g)。滴入后,一边加热回流一边搅拌4.5小时。然后,在常压下蒸馏除去丙酮(175ml)。然后,向50~53℃温度下的反应混合物中加入20℃的水(320ml),通过过滤来回收析出的固体。将得到的晶体(221g)用85~90℃的温水(320ml)洗涤,再用40℃的甲醇(188ml)洗涤。在用温水洗涤之后且用甲醇洗涤之前的晶体的温度为72℃。将得到的晶体通过上下真空滤器过滤来回收,将回收的晶体在80℃、40mmhg的条件下减
压干燥,得到2,3,5,6-四氯-1,4-苯二甲酸二甲酯。关于得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯,其收量:201.7g,纯度98.749重量%,对环境有害的副产物六氯苯为0.8ppm,五氯苯为30ppm。
[0168]
[实施例3]
[0169]
向具有玻璃衬里的反应器中投入丙酮(1015ml)和水(172ml)至丙酮的含水量为15重量%,然后投入2,3,5,6-四氯-1,4-苯二甲酸(批次2,200g)和东京化成工业株式会社制的碳酸钠(94g),将反应混合物升温至57℃,向55~58℃温度下的反应混合物中滴入东京化成工业株式会社制的硫酸二甲酯(189g)。滴入后,一边加热回流一边搅拌4.5小时。然后,在常压下蒸馏除去丙酮(175ml)。然后,向50~53℃温度下的反应混合物中加入20℃的水(320ml),通过过滤来回收析出的固体。将得到的晶体用85~90℃以上的温水(160ml)洗涤,再用40℃的甲醇(188ml)洗涤。在用温水洗涤之后且在用甲醇洗涤之前的晶体的温度为59℃。将得到的晶体通过上下真空滤器过滤来回收,将回收的晶体在80℃、40mmhg的条件下减压干燥,得到2,3,5,6-四氯-1,4-苯二甲酸二甲酯。关于得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯,其收量:201.9g,纯度98.241重量%,对环境有害的副产物六氯苯为8.1ppm,五氯苯为30ppm。洗涤水和甲醇的温度及使用量、用温水洗涤之后且用甲醇洗涤之前的晶体的温度、2,3,5,6-四氯-1,4-苯二甲酸二甲酯的收量、纯度、以及对环境有害的副产物的含量分别示于下表1。
[0170]
[0171][0172]
[实施例4~10、比较例4~8]
[0173]
将洗涤水和甲醇的温度及使用量分别变更为如表1所示,除此以外,与实施例3同
样操作,分别得到实施例4~6和比较例4~6的2,3,5,6-四氯-1,4-苯二甲酸二甲酯。关于得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯,收量、纯度、对环境有害的副产物六氯苯和五氯苯的含量、在用温水洗涤之后且用甲醇洗涤之前的晶体的温度分别如表1所示。
[0174]
另外,将洗涤水和有机溶剂的温度及使用量、以及有机溶剂的种类分别变更为如表2所示,除此以外,与实施例3同样操作,分别得到实施例7~10以及比较例7和8的2,3,5,6-四氯-1,4-苯二甲酸二甲酯。关于得到的2,3,5,6-四氯-1,4-苯二甲酸二甲酯,其收量、纯度、对环境有害的副产物六氯苯和五氯苯的含量、在用温水洗涤之后且用有机溶剂洗涤之前的晶体的温度分别如表2所示。
[0175]
由表1的结果可知,在将晶体用30~100℃的温水洗涤、然后再用30~80℃的有机溶剂洗涤的实施例2~10中,洗涤后的晶体中的六氯苯和五氯苯的含量少,被充分除去。其中可知,尤其是在将晶体用85℃以上的温水洗涤的实施例2~4、7和8中,晶体中的六氯苯和五氯苯的含量进一步减少,能够有效去除这些副产物。在实施例2~10中,认为由于在用温水洗涤后且用有机溶剂洗涤之前的晶体的温度较高,因此可充分除去上述副产物。
[0176]
另一方面可知,在将晶体仅用20℃的水洗涤的比较例4、用20℃的水洗涤后再用30~80℃的有机溶剂洗涤的比较例5、以及用20℃的水洗涤后再用20℃的有机溶剂洗涤的比较例6~8中,洗涤后的晶体中的六氯苯和五氯苯的含量多,不能被充分除去。
[0177]
产业实用性
[0178]
本发明提供:在制造用作农业园艺用除草剂的2,3,5,6-四氯-1,4-苯二甲酸二甲酯时,与以往方法相比减少了六氯苯和五氯苯等对环境有害的副产物的含量,能够有效制造的工业化方法,具有产业实用性。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
