1.本发明属于金属纳米簇的可控制备技术领域,具体涉及一种近红外发光的谷胱甘肽保护金纳米簇的可控制备及其发光性能调控方法。
背景技术:
2.金属纳米簇(mncs)是一类新型的超小尺寸金属纳米粒子,因其独特的光致发光特性(包括发光稳定性好、生物相容性佳、发光范围可调和斯托克斯位移大等优点),使其广泛应用于生物传感和生物成像领域。其中金纳米簇(auncs)是目前研究最广泛和最深入的典型纳米团簇,吸引了人们越来越多的研究兴趣。它们具有典型的核-壳结构,由几个到上百个au原子构成的金属核和表面配体的壳组成。研究表明,auncs的光致发光性能与金属核组成、结构、大小和配体分子结构等密切相关。一般情况下,金纳米簇的光致发光波长随着金属核的增大逐渐红移,能够实现从可见光到近红外光区的调控。由于金纳米簇的原子个数是一组幻数,不连续分布,因此金纳米簇的光致发光波长虽然可调控,但不连续且不可控。
3.金属纳米簇的发光性能与金属核的尺寸、形状和组成密切相关,因此人们通过掺杂其它金属原子来改变金属核的几何和电子结构,并进一步改善它们的热稳定性和光学性质。研究表明通过金属掺杂可以成功地改善和调控金属纳米簇的光致发光波长和发光强度,这极大地拓展了金属纳米簇在生物传感和生物成像等领域的应用。例如:rongchao jin等人通过ag原子掺杂进auncs,使其荧光量子产率提升了200倍
1.。liu等人获得了近红外发射的auncs,并掺杂了ag(i)离子,导致荧光发射猝灭和蓝移
2.。因此,掺杂其它金属原子是一种有效的改善和调控纳米簇发光性能的方法,进一步拓展了金属纳米簇的实际应用范围。
4.尤其是近红外发光的金属纳米簇,由于该光区能够避免生物体内源荧光的干扰、具有更好的光穿透性和减小光散射等对荧光信号的干扰,因此近红外发光的金纳米簇被认为是最有前途的生物传感和生物成像材料。但目前已报道的近红外发光金属纳米簇种类较少、荧光量子效率较低且发光范围不易调控,极大地限制了该类纳米材料的实际应用。本发明采用一种简单、快速的水热合成法,将谷胱甘肽(gsh)作为保护配体,柠檬酸钠为还原剂合成近红外发光的金纳米簇(auncs@gsh)。通过调节金与配体谷胱甘肽的比例、还原剂柠檬酸钠的浓度、反应温度及反应时间等,制备出近红外发光的金纳米簇(auncs@gsh)。此外,通过引入ag(i)离子可以调控金纳米簇的发光范围从805nm的荧光发射峰逐渐转变为红光发射,并使其在615nm~720nm波长范围内发光波长连续可调。
5.[1]wang s,meng x,das a,et al.a 200-fold quantum yield boost in the photoluminescence of silver-doped ag
(x)
au
(25-x)
nanoclusters:the 13th silver atom matters.angew.chem.int.ed.2014,53,2376
–
2380.
[0006]
[2]wang y,liu l,gong l,et al.reactivity toward ag
:a general strategy to generate a new emissive center from nir-emitting gold nanoparticles.j.ph
ys.chem.lett.2018,9,557-562。
技术实现要素:
[0007]
本发明的目的在于提供一种近红外发光金纳米簇的可控制备及其发光性能调控的方法。首先,通过水热合成方法和条件优化(金与配体谷胱甘肽比例、还原剂柠檬酸钠浓度、反应温度、反应时间)可控制备谷胱甘肽保护的金纳米簇(auncs),使其具有优异的近红外发光特性(805nm)。此外,通过掺杂ag(i)离子可以调控该纳米簇的发光范围,使其发射波长从近红外光区逐渐转移到红光区,并在615nm~720nm波长范围内实现荧光发射波长的连续可调。
[0008]
本发明所述的近红外发光金纳米簇的可控制备及其发光性能调控方法,是以氯金酸(haucl4·
3h2o)作为au源,谷胱甘肽(gsh)作为配体分子,柠檬酸钠作为还原剂;其步骤如下:
[0009]
(1)将氯金酸溶液、谷胱甘肽溶液和柠檬酸钠溶液充分混匀,然后再加入蒸馏水,得到混合物溶液(如没有特别强调,本发明所述的溶液均为水溶液);
[0010]
(2)将步骤(1)的混合物溶液再次混匀,并转移至内衬为聚四氟乙烯的反应釜中,在100~120℃条件下反应0.5h~4.0h,得到谷胱甘肽保护的金纳米簇溶液;
[0011]
(3)向步骤(2)得到的谷胱甘肽保护的金纳米簇溶液中加入无水乙醇,振荡混合均匀,然后6000~8000rpm下离心10~20分钟;离心后,去除上清除掉过量的柠檬酸钠和谷胱甘肽;最后冻干沉淀,4℃避光保存,得到近红外发光金纳米簇粉末;
[0012]
(4)将步骤(3)得到的近红外发光金纳米簇粉末加入到10mm、ph=6.5~7.0的mes-naoh缓冲液中,配置成浓度0.04~0.06mg
·
l-1
的金纳米簇溶液,然后向其中逐渐加入硝酸银溶液,硝酸银的终浓度为0.5~120μm;随着硝酸银的加入,auncs的粒径增加且其表面电子能发生变化,从而实现对近红外发光的谷胱甘肽保护金纳米簇的可控制备和荧光发射波长的连续调控。
[0013]
步骤(1)的蒸馏水中氯金酸的终浓度为8mm,氯金酸与gsh摩尔用量比例为1:3~8,氯金酸与柠檬酸钠摩尔用量比例为1:20~50。
[0014]
ag(i)离子诱导该近红外发光金纳米簇的荧光发射波长变化可以分为两阶段,在第一阶段中,ag(i)离子浓度在0.5~20μm时,auncs的发射波长从805nm逐渐转移到615nm,在第二阶段中,ag(i)离子浓度在20~120μm时,发射从615nm逐渐红移到720nm。进一步机理研究表明:第一阶段的比率型荧光变化归因于金纳米簇对ag(i)离子的反氧化还原和金银双金属纳米团簇的形成,并引起发射波长改变和增强;当ag(i)离子含量进一步增加时,该纳米簇金属核的粒径增长和组成变化导致第二阶段的发射峰位红移和猝灭。
附图说明
[0015]
图1:(a)au与gsh之间不同比例时auncs@gsh的荧光发射谱图,(b)au与gsh之间不同比例时对应在805nm处荧光强度的柱状图。
[0016]
图2:(a)不同柠檬酸钠浓度下auncs@gsh荧光发射光谱图,(b)不同柠檬酸钠浓度下对应在805nm处荧光强度的柱状图。
[0017]
图3:(a)在100℃时不同反应时间下auncs@gsh的荧光发射光谱图,(b)在100℃时
不同反应时间下805nm处的强度变化图,(c)在110℃时不同反应时间下auncs@gsh的荧光发射光谱图,(d)在110℃时不同反应时间下805nm处的强度变化图,(e)在120℃时不同反应时间下auncs@gsh的荧光发射光谱图,(f)在120℃时不同反应时间下805nm处的强度变化图,
[0018]
图4:(a)auncs的紫外吸收、激发、发射光谱图,(b)auncs的透射电镜图,(c)auncs的粒径分布图,(d)auncs的金元素的光电子能谱图。
[0019]
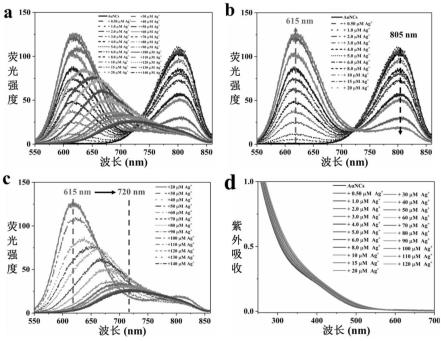
图5:auncs在不同浓度ag(i)离子存在下的荧光光谱((a)0~140μm,(b)0~20μm,(c)20~140μm,(d)auncs在不同浓度ag(i)离子(0~120μm)下的紫外-可见吸收光谱。
[0020]
图6:(a)auncs在不同浓度ag(i)离子下的金的含量和价态分布,(b)auncs与120μm ag(i)离子作用时银的含量和价态分布图。
[0021]
图7:auncs在不同浓度ag(i)下的(a)s 2p,(b)n 1s和(c)o 1s的xps图。
[0022]
图8:auncs在不同浓度ag(i)(0,6.0,20,70,120μm)下的dls图。
[0023]
图9:(a)auncs在6.0μm ag(i)离子下的tem图,(b)auncs在6.0μm ag(i)离子下的粒径统计分布图,(c)auncs在20μm ag(i)离子下的tem图,(d)auncs在20μm ag(i)离子下的粒径统计分布图,(e)auncs在120μm ag(i)离子下的tem图,(f)auncs在120μm ag(i)离子下的粒径统计分布图。
[0024]
图10:(a)auncs中滴定半胱氨酸的荧光光谱,(b)auncs与6.0μm ag(i)离子下滴定半胱氨酸的荧光光谱,(c)auncs与20μm ag(i)离子下滴定半胱氨酸的荧光光谱,(d)auncs与70μm ag(i)离子下滴定半胱氨酸的荧光光谱,(e)auncs与120μm ag(i)离子下滴定半胱氨酸的荧光光谱(cys,0
–
100μm)。
[0025]
图11:(a)auncs中滴定edta的荧光光谱,(b)auncs与6.0μm ag(i)离子下滴定edta的荧光光谱,(c)auncs与20μm ag(i)离子下滴定edta的荧光光谱,(d)auncs与70μm ag(i)离子下滴定edta的荧光光谱,(e)auncs与120μm ag(i)离子下滴定edta的荧光光谱(0
–
100μm)。
[0026]
图1对应实施例1;图2对应实施例2;图3对应实施例3;图4对应实施例4;图5对应实施例5;图6~7对应实施例6;图8~9对应实施例7;图10~11对应实施例8。
[0027]
如图1-4所示,通过水热合成的方法,分别对实验条件如氯金酸与配体谷胱甘肽的比例、还原剂柠檬酸钠的浓度、反应温度、反应时间进行优化,成功制备出的发光最优的近红外金纳米簇(805nm)。并对得到的金纳米簇采取以下方式进行表征:该纳米簇激发波长为440nm,发射波长为805nm,紫外吸收为400nm。采用透射电子显微镜(tem)观察金纳米簇的表面形貌和大小,该纳米簇分散性较高、粒径较为均一,平均粒径为~1.3nm。此外,在xps结果中,auncs在83.8ev和87.5ev存在两个特征峰,它们分别属于au 4f
7/2
和4f
5/2
。对au 4f
7/2
峰进行分峰处理后,可分为结合能为83.8ev和84.8ev的两个峰,分别归于au(0)和au(i)。au(0)与au(i)的所占比例分别为94%和6%,表明在制备过程中au(iii)基本上都被还原成au(0),综合以上结果,表明金纳米簇成功制备。
[0028]
如图5所示,将冻干的纯的金纳米簇粉末配置成浓度为0.050mg
·
l-1
的溶液,再将硝酸银溶液滴定到auncs溶液中,当ag(i)离子浓度在0~20μm时,生成新的发射峰(615nm)并荧光增强,而805nm处荧光逐渐猝灭。此外,当ag(i)离子含量继续增多时,615nm处荧光逐渐猝灭并发射红移,最终在ag(i)离子浓度达到120μm时,红移至720nm并呈现微弱的发光。因此,ag(i)离子的引入可以调控近红外金纳米簇荧光发射。
[0029]
如图6~7所示,深入探究荧光变化原因,通过xps测定各元素在加入银离子前后的氧化态变化,xps结果分析表明,在该过程中,au4f
7/2
的结合能从83.8ev转移到84.2ev,最终达到85.0ev,同时,au(0)的比例从94%下降到64.5%,最后金属核中几乎没有残留的au(0),说明核内au(0)与ag(i)之间发生了氧化还原反应,导致au(0)被氧化成au(i)。以及银以零价形式ag(0)在体系中存在,表明ag(i)离子确实被还原为ag(0),说明了金属核的组成确实发生了变化。在s 2p、n 1s和o1s的xps结果中,它们的结合能几乎保持不变,说明该阶段的荧光变化源于auncs@gsh对ag(i)离子的反氧化还原,并形成金银双金属纳米团簇。
[0030]
如图8~9所示,动态光散射和透射电镜结果显示,随着ag(i)离子浓度的增加,纳米簇的粒径发生增长,说明了该过程的发光增强和红移是由于反氧化还原过程中金属核的粒径增长和组成改变所致。
[0031]
如图10~11所示,为了进一步阐明该过程的起源和动力学变化,将半胱氨酸(cys)和乙二胺四乙酸二钠(edta)引入到ag(i)离子存在下的近红外发射的auncs@gsh中。半胱氨酸使荧光都显著增强,并未恢复到原来的近红外发射,说明该发光变化不可逆。同时,edta也未引起任何荧光变化,说明au-agncs表面没有形成ag(i)-羧酸盐。其中,加入ag(i)离子导致纳米簇表面覆盖度大大降低,而半胱氨酸的引入,增加了表面配体覆盖度从而诱导au-agncs@gsh发光增强。该结果进一步说明ag(i)离子与金核紧密结合,导致粒径增长和表面覆盖度降低。
具体实施方式
[0032]
本发明中使用的三水合氯金酸(haucl4·
3h2o,》99.9%)、谷胱甘肽(gsh)、柠檬酸钠(ca)、2-吗啉乙磺酸(mes)购买于上海阿拉丁生化科技有限公司(中国上海),硝酸银(agno3,》99.9%)购买于中国北京化学试剂公司,氢氧化钠(naoh)购买于国药集团化学试剂有限公司,所有的化学药品均是分析纯,且没有再纯化。超纯水用于整个实验过程。
[0033]
实施例1:
[0034]
通过水热合成法制备谷胱甘肽保护的金纳米簇(auncs):以氯金酸(haucl4·
3h2o)作为au源,谷胱甘肽(gsh)作为配体,柠檬酸钠作为还原剂。首先,探究氯金酸与配体谷胱甘肽的用量比例对金纳米簇发光的影响。先向不锈钢反应釜中加入800μl、10mm氯金酸溶液,然后按照氯金酸与gsh摩尔用量比例1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10分别加入100mm谷胱甘肽溶液160、240、320、400、480、560、640、720、800μl,再加入400μl、500mm柠檬酸钠溶液,然后用蒸馏水补充使反应总体积为10ml;将混合物溶液充分混匀,并将反应釜转移至烘箱中,反应时间2h,反应温度110℃,待反应冷却至室温后,通过荧光光谱测量产物。
[0035]
结果显示:随着gsh浓度的增加,805nm处的荧光发射峰出现并逐渐增强,直到氯金酸与gsh的摩尔用量比例为1:4,然后荧光强度略有下降。因此,选择320μl、100mm gsh作为金纳米簇发射的最佳浓度(图1),此时gsh的终浓度为3.2mm,较为适合的氯金酸与gsh摩尔用量比例为1:3~8。
[0036]
实施例2:
[0037]
接着探究还原剂柠檬酸钠的浓度对金纳米簇发光的影响,向不锈钢反应釜中加入800μl、10mm氯金酸溶液,320μl、100mm谷胱甘肽溶液,分别加入500mm柠檬酸钠溶液80、160、240、320、400、480、560、640、720、800μl,将混合物充分混匀,并将反应釜转移至烘箱,统一
设置反应时间2h,反应温度110℃,待反应冷却至室温后,通过荧光光谱测量产物。
[0038]
结果显示:随着柠檬酸钠浓度的增加,805nm处的荧光发射峰出现并逐渐增强,直到柠檬酸钠终浓度为20mm时,荧光强度略有下降。因此,选择400μl、500mm柠檬酸钠作为金纳米簇发射的最佳浓度(图2),此时氯金酸与柠檬酸钠摩尔用量比例为1:25,较为适合的氯金酸与柠檬酸钠摩尔用量比例为1:20~50(对应柠檬酸钠终浓度为16~40mm的情形)。
[0039]
实施例3:
[0040]
最后探究反应时间和反应温度对金纳米簇的发光的影响,向不锈钢反应釜中加入800μl、10mm氯金酸溶液,320μl、100mm谷胱甘肽溶液,400μl、500mm柠檬酸钠,用蒸馏水补充使反应总体积为10ml,将混合物充分混匀,并将反应釜转移至烘箱,设置反应温度分别为100℃、110℃、120℃,分别加热0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0h,待反应冷却至室温后,通过荧光光谱测量产物。
[0041]
结果显示:在110℃的条件下反应2.5小时获得的auncs@gsh荧光强度最强(图3),在110℃的条件下较为适合的反应时间为2.0~4.0h;
[0042]
实施例4:
[0043]
基于实施例1~3,制备近红外金纳米簇的最佳条件:800μl、10mm氯金酸溶液,320μl、100mm谷胱甘肽溶液,400μl、500mm柠檬酸钠溶液,用蒸馏水补充使反应总体积为10ml,将混合物充分混匀,并将反应釜转移至烘箱,设置反应温度110℃,水热反应2.5h,得到谷胱甘肽保护的金纳米簇溶液。向得到的谷胱甘肽保护的金纳米簇溶液中加入无水乙醇,振荡混合均匀,使用高速离心机7000rpm离心15分钟;离心后,去除上清除掉过量的柠檬酸钠和谷胱甘肽。最后冻干沉淀,4℃避光保存,得到近红外发光金纳米簇粉末。
[0044]
对获得的近红外发光金纳米簇粉末利用荧光光谱、紫外-可见吸收光谱、透射电子显微镜(tem)和x-射线光电子能谱(xps)等方法进一步表征。
[0045]
结果显示:该纳米簇激发波长440nm,发射波长为805nm,在紫外-可见吸收光谱图中观察到一个约在400nm的宽肩峰(图4a),这表明在反应过程中没有产生大的纳米颗粒,该结果与报道的auncs一致。利用透射电子显微镜(tem)观察表面形貌以及大小。auncs呈现高度结晶和单分散的状态,晶格间距为0.23nm,与面心立方au(111)晶面的d间距一致(图4b)。通过对200个粒子统计,大部分auncs粒径分布在0.9~1.70nm之间,其平均粒径为1.30nm(图4c),这与紫外可见吸收结果一致。采取x-射线光电子能谱(xps)测定auncs中金的氧化态,auncs在83.8ev和87.5ev存在两个特征峰,它们分别属于au 4f
7/2
和4f
5/2
。对au 4f
7/2
峰进行分峰处理后,可分为结合能为83.8ev和84.8ev的两个峰(图4d),分别归于au(0)和au(i)。au(0)与au(i)的所占比例分别为94%和6%,表明在制备过程中au(iii)基本上都被还原成au(0)。当au(i)存在于纳米簇表面、au(0)位于核内时,配体gsh与au(i)在表面相互作用以稳定auncs。综合以上结果,表明金纳米簇成功制备。
[0046]
实施例5:
[0047]
基于实施例4得到的近红外发射的金纳米簇粉末置于10mm、ph=6.8的mes-naoh缓冲液中,配置成浓度为0.05mg
·
l-1
的金纳米簇溶液;然后向其中加入终浓度为0.5、1.0、2.0、3.0、4.0、5.0、6.0、8.0、10、15、20、30、40、50、60、70、80、90、100、110、120、130、140μm的硝酸银溶液,并记录荧光变化数据,同时,利用紫外-可见吸收光谱观察auncs对ag(i)离子的响应。
[0048]
结果显示:将硝酸银溶液滴定到auncs溶液中,auncs在805nm处的发射迅速猝灭,并在615nm处产生一个新的发射峰,然后随着ag(i)离子浓度的增加,发射红移并猝灭。准确地说,当ag(i)离子浓度在0~20μm时,产生新的发射峰(615nm)的同时荧光增强,而805nm处荧光逐渐猝灭。此外,当ag(i)离子含量继续增多时,615nm处荧光逐渐猝灭并发射峰位红移,最终在ag(i)离子浓度达到120μm时,红移至720nm。同时,随着ag(i)离子的加入,在325nm和450nm附近的吸收逐渐增加,这表明在与ag(i)相互作用时,auncs的表面电子能发生了变化(图5)。
[0049]
实施例6:
[0050]
基于实施例5中的纳米簇的荧光变化,我们进一步利用xps谱对其中的机理深入研究。将实施例5中的最终溶液冷冻干燥处理后,考察加入ag(i)离子前后auncs@gsh中金银的含量及价态变化,并且分别监测s 2p、n 1s和o1s的xps谱变化。
[0051]
结果显示:在20μm ag(i)离子的存在下,au 4f
7/2
的结合能从83.8ev转移到84.2ev,最终在120μm ag(i)离子时达到最大值85.0ev。同时,当ag(i)离子浓度为20μm时,au(0)的比例从94%下降到64.5%,最后,在ag(i)离子浓度达到120μm时,金属核中几乎没有残留的au(0)。以上结果表明,核内au(0)与ag(i)之间发生了氧化还原反应,导致au(0)被氧化成au(i)。接着,通过分析ag的价态及含量变化,发现ag的两个特征峰分别位于368.2ev和374.0ev,分别指代ag 3d
5/2
和ag 3d
3/2
。结合已经报道的agncs,证实了银以零价形式ag(0)在体系中存在。这些结果表明,在滴定过程中,ag(i)离子确实被还原为ag(0)。金银的含量和价态的变化说明了金属核的组成确实发生了变化。同时,当ag(i)离子存在时,s 2p、n 1s和o1s的结合能几乎保持不变。结合文献报道,发现s原子在纳米团簇表面形成au(i)-gs和/或ag(i)-gs复合物。而氮和氧原子的结果表明它们并没有与au
–
agncs表面的ag(i)离子结合。因此,该阶段的荧光变化源于auncs@gsh对ag(i)离子的反氧化还原,并形成金银双金属纳米团簇。
[0052]
实施例7:
[0053]
基于实施例5中的纳米簇的荧光变化,我们进一步利用动态光散射(dls)来监测在不同浓度ag(i)离子下auncs@gsh的粒径变化。同时,通过透射电镜观察auncs@gsh与终浓度为6.0,20和120μm ag(i)离子相互作用后的粒径和形貌变化。
[0054]
结果显示:在动态光散射图中,纯水中单个auncs的尺寸分布较窄,约为1.9nm,而在120μm ag(i)离子下,尺寸逐渐增长到4.9nm,该结果表明,随着ag(i)离子浓度的增加,纳米簇的水合粒径逐渐变大。在透射电镜图中,在6.0μm ag(i)离子的存在下,与单独分散的auncs(1.30nm)相比,产生了更大的纳米颗粒,其平均粒径为1.90nm。当加入20和120μm ag(i)离子时,纳米簇的平均粒径分别增长到2.09和2.23nm。其粒径增长与dls的结果一致,证实了金纳米簇对ag(i)离子的反氧化还原和金银双金属纳米簇的形成。发射与粒径大小相关以及xps结果表明,本研究中的发光增强和红移是由于反氧化还原过程中金属核的粒径增长和组成改变所致。
[0055]
实施例8:
[0056]
基于实施例5中的纳米簇的荧光变化,我们进一步引入半胱氨酸(cys)和乙二胺四乙酸二钠(edta),考察ag(i)离子与auncs@gsh的作用模式。分别配置10mm的半胱氨酸(cys)和乙二胺四乙酸二钠(edta),再滴加到ag(i)离子存在下的近红外发射的auncs@gsh中,并
利用荧光发射光谱监测该过程。
[0057]
结果显示:将半胱氨酸添加到所有au-agncs@gsh中,发现荧光都显著增强,并未恢复到原来的近红外发射。说明该发光变化不可逆。同时,向au-agncs@gsh中滴加edta,发现荧光强度只是略微波动,所以edta未引起任何荧光变化,说明au-agncs表面没有形成ag(i)-羧酸盐。综上所述,ag(i)离子的引入使纳米簇的粒径增长,而纳米簇的表面配体数目没有改变,故导致其表面覆盖度大大降低,不能提供足够的空间位阻来保护金属核结构,并随着尺寸的进一步增大,使越来越多的银原子暴露在金属核表面。因此,半胱氨酸诱导au-agncs@gsh发光增强可归因于半胱氨酸直接作用于核表面并增加了表面配体覆盖度。该结果进一步说明ag(i)离子与金核紧密结合,导致粒径增长和表面覆盖度降低。
[0058]
总之,本发明通过水热合成法获得了一种近红外发光的金纳米簇,其荧光发射峰位与805nm,平均直径为1.30nm。在ag(i)离子存在下,在第一阶段中,auncs的发射峰从805nm转移到615nm,在第二阶段中其发射峰从615nm逐渐红移到720nm。并对其内在机理进行了深入研究,第一阶段归因于金纳米簇对ag(i)离子的反氧化还原和金银双金属纳米团簇的形成,并引起发光增强。当ag(i)离子含量增加时,金属核的粒径增长和组成变化导致第二阶段的发射红移和猝灭。
[0059]
还需要说明的是,本发明的具体实施例只是用来示例性说明,并不以任何方式限定本发明的保护范围,本领域的相关技术人员可以根据上述一些说明加以改进或变化,但所有这些改进和变化都应属于本发明权利要求的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。