1.本发明涉及中药制剂,尤其涉及一种川芎饮片的炮制方法、温经汤及其制备方法。
背景技术:
2.温经汤出自宋陈自明《妇人大全良方》(宋
·
陈自明),其记载了温经汤剂量为当归、川芎、芍药、桂心、牡丹皮、莪术各半两,人参、甘草、牛膝各一两,即当归、川芎、芍药、桂心、牡丹皮、莪术各20.60g,人参、甘草、牛膝各41.30g。温经汤具有温经散寒、通利血脉、活血化瘀、气血双补、肝脾兼调的功效。
3.在对于温经汤的研究过程中发现,制备温经汤所用的原药材,采用不同的炮制方式,会影响其最终有效成分的溶出,进而影响制备的温经汤的品质。
4.川芎的炮制方法是将川芎原药材去杂质,分档,略浸,洗净,润透,切薄片,晒干或低温干燥制得,但川芎的主要成分阿魏酸在湿、热条件下稳定性较差,因此炮制过程中润透用水量、时间和干燥的温度、时间等因素都对川芎饮片中阿魏酸含量有着明显的影响,如何在符合中药饮片要求的前提下,炮制出具有较高阿魏酸含量的川芎饮片,一直是对于川芎炮制的研究重点。
5.当归的主要有效成分也包括阿魏酸,在当归的炮制过程中也需要综合考虑温度、时间和水量等因素。
6.芍药的炮制过程中同样存在着有效成分易损失的问题,芍药的主要成分芍药苷易水解,同时对温度较为敏感,因此炮制过程中润透用水量、时间和干燥的温度、时间等因素都对芍药中芍药苷含量也有着明显的影响。
7.牡丹皮的炮制过程中也存在着有效成分易损失的问题,牡丹皮的主要成分丹皮酚挥发性强、水溶性差,在炮制过程中防止丹皮酚挥发对于保留牡丹皮中的有效成分有着明显影响。
8.人参皂苷是人参中的主要有效成分,其包括人参皂苷rb1、人参皂苷re和人参皂苷rg1等,这三种人参皂苷在热环境下会发生不同程度的降解反应,因此在炮制过程中,如何在符合中药饮片要求的前提下,最大程度的保留人参中的有效成分也是炮制的难点之一。
9.牛膝在贮藏过程中易发生“泛糖”现象,是最容易变质的药材之一。其中,水分和温度都是影响牛膝“泛糖”的主要因素。而牛膝“泛糖”也进一步影响着其有效成分蜕皮甾酮的含量。因此在牛膝炮制过程中,控制水分和温度,将影响制备的牛膝饮片的质量和蜕皮甾酮含量。
技术实现要素:
10.针对上述问题,本发明提供一种川芎饮片的炮制方法、温经汤及其制备方法。
11.为实现上述目的,本发明所采用的技术方案为:
12.一种川芎饮片的炮制方法,是取川芎原药材净制后,加入川芎原药材重量0.4~0.6倍的水,闷润1.9~2.1h,再经切片,60
±
2℃干燥1.4~1.6h,即得所述川芎饮片。
13.一种利用上述川芎饮片作为原料制成的温经汤,制成所述温经汤的有效成分的原料,还包括当归、芍药、桂心、牡丹皮、莪术、人参、甘草和牛膝;
14.制成所述单服温经汤的原料用量为当归、川芎饮片、芍药、桂心、牡丹皮、莪术各1.67g重量份,人参、甘草、牛膝各3.33g重量份;
15.所述芍药的炮制方法是取芍药原药材净制后,加入芍药原药材重量0.9~1.1倍的水,闷润1.9~2.1h,再经切片,60
±
2℃干燥1.4~1.6h制得。
16.进一步的,所述牡丹皮的炮制方法是取牡丹皮原药材净制后,加入牡丹皮原药材重量0.4~0.6倍的水,闷润2.9~3.1h,再经切片,60
±
2℃干燥0.9~1.1h制得。
17.进一步的,所述人参的炮制方法是取人参原药材净制后,加入人参原药材重量0.4~0.6倍的水,闷润1.9~2.1h,再经切片,60
±
2℃干燥2.4~2.6h制得。
18.进一步的,所述当归的炮制方法是取当归原药材净制后,加入当归原药材重量0.31~0.35倍的水,闷润1.4~1.6h,再经切片,60
±
2℃干燥0.9~1.1h,所得当归切片,加8~10%vol黄酒拌匀,闷透,炒制5
±
0.5min,取出,室温放凉后制得;
19.其中当归切片与黄酒的重量比为10:0.9~1.1。
20.进一步的,所述牛膝的炮制方法是取牛膝段,加8~10%vol黄酒拌匀,闷透,文火炒至表面干燥(酒炒时间为8min),易折断时,取出,放凉制得;
21.其中牛膝段与黄酒的重量比为10:0.9~1.1。
22.所述甘草的炮制方法是取甘草原药材净制后,加入甘草原药材重量0.4~0.6倍的水,闷润2.9~3.1h,再经切片,文火炒制110~130s制得。
23.一种温经汤的制备方法,是取当归、川芎饮片、芍药、桂心、牡丹皮、莪术、人参、甘草和牛膝经粉碎过筛后,加水、加盖,浸泡30min直接依次进行武火煎煮和文火煎煮,再经过滤,所得药液直接进行冻干,即得所述温经汤。
24.进一步的,所述冻干的过程包括预冻和升华干燥;
25.所述预冻的温度为-40℃、时间为16h;
26.所述升华干燥的温度为-49℃、时间为96h;
27.无需解析干燥。
28.进一步的,所述武火煎煮的时间为14~16min;文火煎煮的时间为59~61min。
29.本发明的川芎饮片的炮制方法、温经汤及其制备方法的有益效果为:
30.本发明通过改变炮制过程中的工艺参数,最大程度的保留了川芎饮片中阿魏酸的含量,提高川芎饮片的药效;
31.本发明利用川芎饮片制备的温经汤,能够最大程度的保留有效成分,防止有效成分挥发或分解,有利于提高温经汤中的有效成分含量,进而提高温经汤的品质;
32.本发明通过改变炮制芍药的工艺参数,有效防止了芍药苷的水解,最大程度的保留了芍药中芍药苷的含量;
33.本发明通过改变炮制牡丹皮的工艺参数,有效防止了丹皮酚的挥发,最大程度的保留了牡丹皮中丹皮酚的含量;
34.本发明通过改变炮制人参的工艺参数,寻找到三种人参皂苷的降解程度最低的炮制方法,最大程度的保留了三种人参皂苷的总含量;
35.本发明通过改变炮制牛膝的工艺参数,有效防止了牛膝“泛糖”现象的发生,且最
大程度的保留了蜕皮甾酮的含量,同时也保证了炮制所得的牛膝的品质;
36.本发明通过改变不同原药材的炮制工艺,最大程度的保留了各味药材中所含有效成分,提高有效成分的溶出,进而提高温经汤的品质;
37.本发明通过调整温经汤的制备工艺,将其制成冻干粉,便于储存、服用和工业化生产;
38.本发明通过调整温经汤制备的工艺参数,能够最大程度的保留其中所含有效成分,且能够充分保留其中的不稳定性成分,提高产品品质。
附图说明
39.图1是本发明实验例1中川芎炮制过程中闷润时间考察对比图;
40.图2是本发明实验例1中芍药炮制过程中水用量考察对比图;
41.图3是本发明实验例1中芍药炮制过程中闷润时间考察对比图;
42.图4是本发明实验例1中牡丹皮炮制过程中水用量考察对比图;
43.图5是本发明实验例1中牡丹皮炮制过程中闷润时间考察对比图;
44.图6是本发明实验例1中人参炮制过程中水用量考察对比图;
45.图7是本发明实验例1中人参炮制过程中闷润时间考察对比图;
46.图8是本发明实验例1中当归炮制过程中水用量考察对比图;
47.图9是本发明实验例1中当归炮制过程中闷润时间考察对比图;
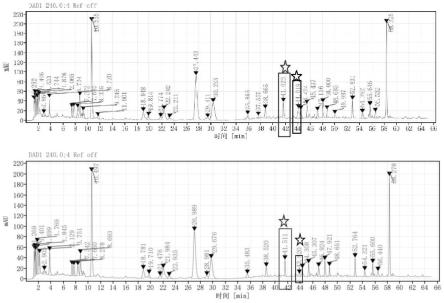
48.图10是本发明实验例1中温经汤冻干粉m1复溶后药液测定的一组超高效液相色谱图;
49.图11是本发明实验例1中温经汤冻干粉d6复溶后药液测定的一组超高效液相色谱图;
50.图12是本发明实验例1中温经汤冻干粉d7复溶后药液测定的一组超高效液相色谱图;
51.图13是本发明实验例1中温经汤冻干粉m1和d7复溶后药液测定的超高效液相色谱图挥发性成分对比图。
具体实施方式
52.下面对本发明实施例中的技术方案进行清楚、完整地描述。在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
53.实施例1一种温经汤的制备方法
54.本实施例为一种温经汤的制备方法,具体制备过程包括依次进行的以下步骤:
55.1)川芎饮片的炮制方法
56.取100g川芎原药材除去杂质后,分开大小,洗净,加入50g水拌匀,闷润2h,此时所得闷润川芎的横截面刚好润透、无干心,再将闷润川芎取出切厚片(2~4mm),放入烘箱中以60
±
2℃进行干燥2h,即得川芎饮片,标记为cx1。
57.2)芍药的炮制方法
58.取100g四川中江白芍药原药材除去杂质后,分开大小,洗净,加入100g水拌匀,闷润2h,此时所得闷润芍药的横截面刚好润透、无干心,再将闷润芍药取出切厚片(2~4mm),放入烘箱中以60
±
2℃进行干燥1.5h,即得芍药(也就是芍药饮片,标记为sy1)。
59.3)牡丹皮的炮制方法
60.取100g陕西商洛景牡丹皮原药材除去杂质后,分开大小,洗净,加入50g水闷润3h,此时所得闷润牡丹皮的横截面刚好润透、无干心,再将闷润牡丹皮取出切薄片(1~3mm),放入烘箱中以60
±
2℃进行干燥1h,即得牡丹皮(也就是牡丹皮饮片,标记为md1)。
61.4)人参的炮制方法
62.取100g吉林长白山净人参原药材除去杂质后,分开大小,洗净,加入50g水闷润2h,此时所得闷润人参的横截面刚好润透、无干心,再将闷润人参取出切薄片(1~3mm),放入烘箱中以60
±
2℃进行干燥2.5h,即得人参(也就是人参饮片,标记rs1)。
63.5)牛膝的炮制方法
64.取牛膝段100g,加入10g度数为9%vol的黄酒拌匀,闷透,置炒制容器内,用文火220w炒至表面干燥(酒炒时间为8min),易折断时,取出,放凉,即得牛膝(也就是牛膝饮片,标记nx1)。
65.7)当归的炮制方法
66.取100g甘肃岷县净当归原药材除去杂质后,分开大小,洗净,加入33.3g水闷润1.5h,所得闷润当归如图7中7b所示,此时所得闷润当归的横截面刚好润透、无干心,再将闷润当归取出切薄片(1~3mm),放入烘箱中以60
±
2℃进行干燥1h,所得当归切片,再加9%vol黄酒拌匀,闷透,置铁锅内,用电磁炉180w炒5min,取出,室温放凉,即得当归(也就是当归饮片,标记dg1)。其中,当归切片与黄酒的重量比为10:1。
67.7)甘草的炮制方法
68.取一定质量的甘草,喷洒一定质量的水,水/甘草药材约为20-23%,密封,60℃条件下软化,以甘草折90
°
而不折为闷润合格,切片,用文火将甘草炒黄为度。
69.8)莪术的炮制方法
70.取莪术原药材按照2005年《浙江省中药饮片炮制规范》标准执行中公开的“醋炒”炮制工艺进行炮制。
71.9)桂心的炮制方法
72.取肉桂原药材按照2015版《中国药典》中公开的炮制工艺进行炮制。
73.10)温经汤的制备
74.取当归、川芎饮片、芍药、桂心、牡丹皮、莪术各1.67g,人参、甘草、牛膝各3.33g,进行药材粉碎通过1号筛,不筛除细粉,加至陶瓷容器中,再加入450ml水,加盖,浸泡30min后直接用武火煎煮15min,文火煎煮60min,此时剩余水量为240ml,煎煮完成后采用100目筛网过滤(过滤用时8s,所得药液直接进行真空冷冻干燥(冻干),真空冷冻干燥分为预冻和升华干燥;
75.预冻:将药液移至-40℃冰箱内,降温至-40℃,维持-40℃预冻16h;
76.升华干燥:抽真空使箱内压力为12pa,以-55℃进行升华干燥72h;
77.干燥完成后,即得温经汤(也就是温经汤冻干粉,标记为m1)。
78.实施例2~6温经汤的制备方法
79.实施例2~6分别为一种温经汤的制备方法,它们的步骤与实施例1基本相同,不同之处仅在于工艺参数的不同,具体详见表1:
80.表1实施例2~6中各项工艺参数一览表
[0081][0082][0083]
实施例2~6其它部分的工艺步骤和参数与实施例1相同,达到的效果也基本与实施例1相同,因此不再赘述。
[0084]
实验例1对照实验
[0085]
一)川芎炮制的对照实验
[0086]
11)水用量考察
[0087]
取100g川芎原药材除去杂质后,分开大小,洗净,分别加入33.3g水(川芎原药材与水重量比3:1)、100g水(川芎原药材与水重量比1:1)拌匀,闷润2h,与实施例1中所得闷润川芎进行对比,川芎原药材与水重量比3:1时,川芎原药材仅表皮浸润,切开有白心;川芎原药材与水重量比1:1时,川芎原药材完全润通,水过量未吸净;而实施例1中川芎原药材与水重量比2:1时,川芎原药材完全润透,水可完全吸净。
[0088]
12)闷润时间考察
[0089]
取100g川芎原药材除去杂质后,分开大小,洗净,加入50g水拌匀,分别闷润1h、3h,与实施例1中所得闷润川芎进行对比,见图1,可以看出,闷润1h时,见图1中1a,所得闷润川芎切开后可见白心,易炸心,碎末较多;闷润3h时,见图1中1c,闷润时间过长,导致闷润川芎表面稍干;而实施例1中闷润2h时,见图1中1b,川芎原药材刚好润透,切开无白心。
[0090]
13)干燥时间考察
[0091]
净川芎100g,洗净,加水50g拌匀,闷润2h后取出切厚片(2~4mm),放入烘箱中以60℃进行干燥,以0.5h、1h、1.5h、2h、2.5h、3h六个时间水平进行川芎饮片的干燥时间考察。
[0092]
观察不同水平下得到的川芎饮片性状并测定川芎饮片中有效成分阿魏酸含量,以确定最优干燥时间。
[0093]
所得川芎饮片性状见表2。
[0094]
表2川芎饮片性状的干燥时间考察结果
[0095][0096][0097]
再次取干燥0.5h、1h、1.5h、2h、2.5h、3h后,所得川芎饮片分别进行有效成分含量测定,以判定该干燥温度和时间是否会对有效成分造成影响,具体实验如下:
[0098]
精密称定1g川芎饮片的粉末(过三号筛),置于具塞锥形瓶中,精密加入30ml甲醇,密塞,称定重量,超声处理30分钟(功率300w,频率40khz),滤过,用10ml甲醇分数次洗涤容器及残渣,合并滤液和洗液,蒸干,残渣加甲醇使溶解,转移至5ml量瓶中,加甲醇至刻度,摇匀,经0.22μm微孔滤膜过滤,即得川芎供试品溶液。
[0099]
精密称定9.95mg阿魏酸对照品,置于50ml容量瓶中,加浓度为70wt%的甲醇水溶液定容至刻度,摇匀,过0.22μm微孔滤膜,得阿魏酸对照品溶液。
[0100]
取阿魏酸对照品溶液和川芎供试品溶液分别采用超高效液相色谱法进行测定,超高效液相色谱法的测定条件为:
[0101]
以甲醇-0.1wt%醋酸水溶液(30:70)为流动相;检测波长为321nm;流速为0.8ml/min;柱温35℃;进样量10μl;在该色谱条件下阿魏酸峰与其他成分峰有较好的分离。理论塔板数按阿魏酸峰计算不低于4000。
[0102]
具有有效成分含量测定结果见表3。
[0103]
表3干燥不同时间所得川芎饮片中阿魏酸的含量一览表
[0104][0105][0106]
表2可以看出,由饮片干燥程度判断,川芎饮片60℃下干燥1.5h时,干燥程度为佳。表3可以看出不同干燥时间,所得川芎饮片中阿魏酸的数据之间p<0.05,差别具有统计学意义,其中干燥1.5h时,阿魏酸含量最高,说明60℃下干燥1.5h可以最大程度的保留阿魏酸。结合表2和表3的数据结果可知,本发明实施例1中的干燥条件最佳,即可以达到干燥程度最佳的效果,又有利于最大程度的保留阿魏酸,防止阿魏酸分解。
[0107]
二)芍药炮制的对照实验
[0108]
21)水用量考察
[0109]
取100g四川中江白芍药原药材除去杂质后,分开大小,洗净,分别加入50g水(芍药原药材与水重量比2:1)、200g水(芍药原药材与水重量比1:2)拌匀,闷润2h,与实施例1中所得闷润芍药进行对比,见图2,可以看出,芍药原药材与水重量比2:1时,见图2中2a,芍药原药材仅表皮浸润,不易切开,切开有干心;芍药原药材与水重量比1:2时,见图2中2c,芍药原药材完全润透,水过量未吸净;而实施例1中芍药原药材与水重量比1:1时,见图2中2b,芍药原药材完全润透,水可完全吸净。
[0110]
22)闷润时间考察
[0111]
取100g四川中江白芍药原药材除去杂质后,分开大小,洗净,加入100g水拌匀,分别闷润1h、3h,与实施例1中所得闷润芍药进行对比,见图3,可以看出,闷润1h时,见图3中3a,所得闷润芍药不易切开,切开后可见干心,易炸心,饮片厚薄不均;闷润3h时,见图3中3c,闷润时间过长,导致闷润芍药药材软烂,不易成型,粉末较多;而实施例1中闷润2h时,见图3中3b,芍药原药材刚好润透,切开无干心。
[0112]
23)干燥时间考察
[0113]
取净白芍100g,洗净,加水100g拌匀,闷润2h后取出切厚片(2~4mm),放入烘箱中以60℃干燥温度进行干燥,以0.5h、1h、1.5h、2h、2.5h五个时间水平进行白芍饮片的干燥。
[0114]
观察不同水平下得到的白芍饮片性状并测定白芍饮片中有效成分芍药苷含量,以确定最优干燥时间。
[0115]
所得白芍饮片性状见表4。
[0116]
表4白芍饮片性状的干燥时间考察结果
[0117]
干燥时间(h)干燥程度0.5饮片尚未完全干燥完毕,表面可见水渍。1饮片尚未完全干燥完毕,切开后切面可见少量水渍。1.5呈类圆形的薄片。表面淡类白色。切面微带类白色。2饮片外表皮呈黄色,质地较脆。2.5饮片外表皮呈深黄色、极脆。
[0118]
再次取干燥0.5h、1h、1.5h、2h、2.5h后,所得芍药饮片分别进行有效成分含量测
定,具体实验如下:
[0119]
精密称定1g芍药饮片的粉末(过三号筛),置于50ml容量瓶中,加入35ml乙醇,超声处理30分钟(功率240w,频率45khz),放冷,加乙醇至刻度,摇匀,滤过,再经0.22μm微孔滤膜过滤,即得芍药供试品溶液。
[0120]
精密称定10.23000mg芍药苷对照品,置于10ml容量瓶中,加甲醇定容至刻度,摇匀,得母液;精密移取600μl母液置于10ml容量瓶,加甲醇稀释至刻度,摇匀,过0.22μm微孔滤膜,得芍药苷对照品溶液(浓度为60μg/ml)。
[0121]
取芍药苷对照品溶液和芍药供试品溶液分别采用超高效液相色谱法进行测定,超高效液相色谱法的测定条件为:
[0122]
以乙腈-0.1wt%磷酸水溶液(14:86)为流动相;检测波长为230nm;流速为0.8ml/min;柱温25℃;进样量10μl;在该色谱条件下芍药苷峰与其他成分峰有较好的分离。理论塔板数按芍药苷峰计算不低于2000。
[0123]
具有有效成分含量测定结果见表5。
[0124]
表5干燥不同时间所得芍药饮片中芍药苷的含量一览表
[0125]
干燥时间(h)芍药苷含量(%)0.51.5511.631.51.8521.552.51.48
[0126]
表4可以看出,以干燥程度为考察指标,初步认为白芍饮片干燥1.5h为佳。表5可以看出不同干燥时间,所得芍药饮片中芍药苷的数据之间p<0.05,差别具有统计学意义,其中干燥1.5h时,芍药苷含量最高。结合表2和表3的数据结果,说明本发明实施例1中的干燥的干燥条件最佳,即可以达到干燥程度最佳的效果,又有利于最大程度的保留芍药苷,防止芍药苷水解。
[0127]
三)牡丹皮炮制的对照实验
[0128]
31)水用量考察
[0129]
取100g陕西商洛景牡丹皮原药材除去杂质后,分开大小,洗净,分别加入33.3g水(牡丹皮原药材与水重量比3:1)、100g水(牡丹皮原药材与水重量比1:1)拌匀,闷润3h,与实施例1中所得闷润牡丹皮进行对比,见图6,可以看出,牡丹皮原药材与水重量比3:1时,见图4中4a,牡丹皮原药材仅表皮浸润,切开易炸心;牡丹皮原药材与水重量比1:1时,见图4中4c,牡丹皮原药材完全润透,水过量未吸净;而实施例1中牡丹皮原药材与水重量比2:1时,见图4中4b,牡丹皮原药材完全润透,水可完全吸净。
[0130]
32)闷润时间考察
[0131]
取100g牡丹皮原药材除去杂质后,分开大小,洗净,加入50g水拌匀,分别闷润2h、4h,与实施例1中所得闷润牡丹皮进行对比,见图5,可以看出,闷润2h时,见图5中5a,所得闷润牡丹皮切开后可见干心,易炸心,碎末较多;闷润4h时,见图5中5c,闷润时间过长,导致闷润牡丹皮药材软烂,不易成型;而实施例1中闷润3h时,见图5中5b,牡丹皮原药材刚好润透,切开无干心。
[0132]
33)干燥时间考察
[0133]
取净牡丹皮100g,洗净,加水50g拌匀,闷润3h后取出切薄片(1~3mm),放入烘箱中以60℃干燥温度进行干燥,以干燥时间作为考察因素,确定0.25h、0.5h、0.75h、1h、1.25h、1.5h六个时间水平进行牡丹皮饮片的干燥。
[0134]
观察不同水平下得到的牡丹皮饮片性状并测定牡丹皮饮片中有效成分阿魏酸含量,以确定最优干燥时间。
[0135]
所得牡丹皮饮片性状见表6。
[0136]
表6牡丹皮饮片性状的干燥时间考察结果
[0137]
干燥时间(h)干燥程度0.25饮片尚未完全干燥完毕,表面可见水渍。0.5饮片尚未完全干燥完毕,表面可见少量水渍。0.75饮片表面干透,切开可见湿芯。1饮片呈空心圆形或卷曲形干燥薄片。1.25饮片呈空心圆形或卷曲形干燥薄片,质地较脆。1.5饮片呈暗褐色,质地脆、易碎。
[0138]
再次取干燥0.25h、0.5h、0.75h、1h、1.25h、1.5h后,所得牡丹皮饮片分别进行有效成分含量测定,以判定该干燥温度和时间是否会对有效成分造成影响,具体实验如下:
[0139]
精密称定1g牡丹皮饮片的粉末(过三号筛),置于50ml容量瓶中,加入50ml浓度为70wt%甲醇水溶液,密塞,超声处理30分钟(功率240w,频率45khz),放冷,加70wt%甲醇水溶液至刻度,摇匀,过滤,再次过0.22μm微孔滤膜,即得牡丹皮供试品溶液。
[0140]
精密称定9.95mg丹皮酚对照品,置于100ml容量瓶中,加甲醇定容至刻度,过0.22μm微孔滤膜,得丹皮酚对照品溶液(浓度为0.1mg/ml)。
[0141]
取丹皮酚对照品溶液和牡丹皮供试品溶液分别采用超高效液相色谱法进行测定,超高效液相色谱法的测定条件为:
[0142]
以乙腈-水(30:70)为流动相;检测波长为274nm;流速为0.8ml/min;柱温25℃;进样量10μl;在该色谱条件下丹皮酚峰与其他成分峰有较好的分离。理论塔板数按丹皮酚峰计算不低于5000。
[0143]
具有有效成分含量测定结果见表7。
[0144]
表7干燥不同时间所得牡丹皮饮片中丹皮酚的含量一览表
[0145]
干燥时间(h)丹皮酚含量(%)0.251.610.51.660.751.7711.791.251.651.51.61
[0146]
表6可以看出,以干燥程度为考察指标,初步认为牡丹皮饮片干燥1h为佳。表7可以看出不同干燥时间,所得牡丹皮饮片中丹皮酚的数据之间p<0.05,差别具有统计学意义,其中干燥1h时,丹皮酚含量最高。结合表2和表3的数据结果可知,说明本发明实施例1中的
干燥时间有利于最大程度的保留丹皮酚,防止丹皮酚挥发。
[0147]
四)人参炮制的对照实验
[0148]
41)水用量考察
[0149]
取100g吉林长白山净人参原药材除去杂质后,分开大小,洗净,分别加入33.3g水(人参原药材与水重量比3:1)、100g水(人参原药材与水重量比1:1)拌匀,闷润2h,与实施例1中所得闷润牡丹皮进行对比,见图6,可以看出,人参原药材与水重量比3:1时,见图6中6a,人参原药材仅表皮浸润,切开有白心;人参原药材与水重量比1:1时,见图6中6c,人参原药材完全润透,水过量未吸净;而实施例1中人参原药材与水重量比2:1时,见图6中6b,人参原药材完全润透,水可完全吸净。
[0150]
42)闷润时间考察
[0151]
取100g人参原药材除去杂质后,分开大小,洗净,加入50g水拌匀,分别闷润1h、3h,与实施例1中所得闷润人参进行对比,见图7,可以看出,闷润1h时,见图7中7a,所得闷润人参切开后可见白心,易炸心,碎末较多;闷润3h时,见图7中7c,闷润时间过长,导致闷润人参药材软烂,不易成型;而实施例1中闷润2h时,见图7中7b,人参原药材刚好润透,切开无白心。
[0152]
43)干燥时间考察
[0153]
取净人参100g,洗净,加水50g拌匀,闷润2h后取出切薄片(1~3mm),放入烘箱中以60℃干燥温度进行干燥,以干燥时间作为考察因素,确定1h、1.5h、2h、2.5h、3h、3.5h六个时间水平进行人参饮片的干燥。观察不同考察水平下得到的人参饮片性状以确定最优干燥时间。
[0154]
观察不同水平下得到的人参饮片性状并测定人参饮片中有效成分三种人参皂苷总含量,以确定最优干燥时间。
[0155]
所得人参饮片性状见表8。
[0156]
表8人参饮片性状的干燥时间考察结果
[0157]
干燥时间(h)干燥程度1饮片尚未完全干燥完毕,表面可见水渍。1.5饮片尚未完全干燥完毕,表面可见少量水渍。2饮片表面干透,切开可见湿芯。2.5呈圆形或类圆形薄片。外表皮灰黄色。切面淡黄白色或类白色。3饮片呈圆形或类圆形薄片,外表皮灰黄色,质地较脆。3.5饮片呈暗黄色,质地脆、易碎。
[0158]
再次取干燥1h、1.5h、2h、2.5h、3h、3.5h后,所得川芎饮片分别进行有效成分含量测定,以判定该干燥温度和时间是否会对有效成分造成影响,具体实验如下:
[0159]
精密称定1g人参饮片的粉末(过三号筛),置于具塞锥形瓶中,加30ml甲醇,密塞,超声处理30分钟(功率300w,频率40khz),过滤,用10ml甲醇分数次洗涤容器及残渣,合并滤液和洗液,蒸干,残渣加甲醇使溶解,转移至5ml量瓶中,加甲醇至刻度,摇匀,再次过0.22μm微孔滤膜,即得人参供试品溶液。
[0160]
精密称定9.98mg人参皂苷rb1、10.03mg人参皂苷re、11.02mg人参皂苷rg,置于50ml容量瓶中,加甲醇定容至刻度,过0.22μm微孔滤膜,得人参皂苷对照品溶液。
[0161]
取人参皂苷对照品溶液和人参供试品溶液分别采用超高效液相色谱法进行测定,超高效液相色谱法的测定条件为:
[0162]
以乙腈为流动相a,水为流动相b,进行梯度洗脱;检测波长为203nm;流速为0.8ml/min;柱温25℃;进样量10μl;在该色谱条件下人参皂苷峰与其他成分峰有较好的分离。理论塔板数按人参皂苷峰计算不低于6000,其中梯度洗脱程序见表9,具有有效成分含量测定结果见表10。
[0163]
表9梯度洗脱程序(人参)
[0164]
时间(分钟)流速流动相a(%)流动相b(%)00.81981350.81981550.82971700.829711000.840601050.819811200.81981
[0165]
表10干燥不同时间所得人参饮片中人参皂苷的含量一览表
[0166][0167]
表8可以看出,根据人参饮片干燥程度判断,60℃下干燥2.5h为佳。表10可以看出不同干燥时间,所得人参饮片中不同干燥时间的三种人参皂苷的数据之间p<0.05,差别具有统计学意义,其中干燥2.5h时,人参皂苷rb1和人参皂苷re rg1含量最高。结合表8和表10的数据结果可知,说明本发明实施例1中的干燥时间有利于最大程度的保留三种人参皂苷总含量,防止人参皂苷降解。
[0168]
五)牛膝炮制的对照实验
[0169]
51)酒炒时间考察
[0170]
取牛膝段,加黄酒拌匀,比例为10:1,闷透,置炒制容器内,用文火220w分别炒4min、6min、8min、10min(炒制4~10min时,牛膝饮片几乎都可达到表面干燥的效果),取出,室温放凉,所得牛膝饮片分别进行有效成分含量测定,具体实验如下:
[0171]
精密称定1g牛膝饮片的粉末(过三号筛),置于具塞锥形瓶中,加35ml水饱和的正丁醇,密塞,浸泡过夜,超声处理30分钟(功率300w,频率40khz),滤过,用10ml甲醇分数次洗涤容器及残渣,合并滤液和洗液,蒸干,残渣加甲醇使溶解,转移至5ml量瓶中,加甲醇至刻度,摇匀,过0.22μm微孔滤膜,即得牛膝供试品溶液。
[0172]
精密称定9.95mg的β-蜕皮甾酮对照品,置于100ml容量瓶中,加甲醇定容至刻度,过0.22μm微孔滤膜,得蜕皮甾酮对照品溶液(浓度为0.1mg/ml)。
[0173]
取蜕皮甾酮对照品溶液和牛膝供试品溶液分别采用超高效液相色谱法进行测定,
超高效液相色谱法的测定条件为:
[0174]
以乙腈-0.1wt%甲酸水溶液(14:86)为流动相;检测波长为250nm;流速为0.8ml/min;柱温40℃;进样量10μl;在该色谱条件下β-蜕皮甾酮峰与其他成分峰有较好的分离。理论塔板数按β-蜕皮甾酮峰计算不低于4000。
[0175]
具有有效成分含量测定结果见表11。
[0176]
表11酒炒不同时间所得牛膝饮片中β-蜕皮甾酮含量一览表
[0177]
酒炒时间(min)β-蜕皮甾酮含量(%)40.0760.0780.08100.07
[0178]
由表11可以看出不同干燥时间,所得牛膝饮片中β-蜕皮甾酮的数据之间p<0.05,差别具有统计学意义,其中酒炒8min时,β-蜕皮甾酮含量最高,说明本发明实施例1中的酒炒时间有利于最大程度的保留β-蜕皮甾酮。
[0179]
六)当归炮制的对照实验
[0180]
61)水用量考察
[0181]
取100g甘肃岷县净当归原药材除去杂质后,分开大小,洗净,分别加入20g水(当归原药材与水重量比5:1)、100g水(当归原药材与水重量比1:1)拌匀,闷润1.5h,与实施例1中所得闷润当归进行对比,见图8,可以看出,当归原药材与水重量比5:1时,见图8中8a,当归原药材仅表皮浸润,切开易炸心;当归原药材与水重量比1:1时,见图8中8c,当归原药材完全润透,水过量未吸净;而实施例1中当归原药材与水重量比3:1时,见图8中8b,当归原药材完全润透,水可完全吸净。
[0182]
62)闷润时间考察
[0183]
取100g当归原药材除去杂质后,分开大小,洗净,加入33.3g水拌匀,分别闷润1h、2h,与实施例1中所得闷润当归进行对比,见图9,可以看出,闷润1h时,见图9中9a,所得闷润当归切开后可见干心,易炸心,碎末较多;闷润2h时,见图9中9c,闷润时间过长,导致闷润当归药材软烂,不易成型;而实施例1中闷润1.5h时,见图9中9b,当归原药材刚好润透,切开无干心。
[0184]
以切开无干心,药材恰好润透为标准,进行当归炮制中闷润时间的考察,确定闷润时间应为1.5h。综上所述,当归炮制应以药材:水=3:1加水后闷润1.5h进行药材闷润。
[0185]
63)干燥时间与酒炒考察
[0186]
取所得闷润当归切薄片(1~3mm),放入烘箱中以60
±
2℃干燥1h的当归切片,加黄酒拌匀,闷透,置铁锅内,用电磁炉180w炒3min、5min、7min,取出,室温放凉,得当归饮片。
[0187]
分别对所得当归饮片进行有效成分含量测定,具体实验如下:
[0188]
精密称定1g当归饮片的粉末(过三号筛),置于具塞锥形瓶中,加20ml浓度为70wt%甲醇水溶液,密塞,超声处理30分钟(功率300w,频率40khz),放冷,加稀乙醇至刻度,摇匀,过滤,再次过0.22μm微孔滤膜,即得当归供试品溶液。
[0189]
精密称定10.0100mg阿魏酸对照品,置于100ml容量瓶中,加甲醇定容至刻度,过0.22μm微孔滤膜,得阿魏酸对照品溶液(浓度为0.1mg/ml)。
[0190]
取阿魏酸对照品溶液和当归供试品溶液分别采用超高效液相色谱法进行测定,超高效液相色谱法的测定条件为:
[0191]
以乙腈-0.085wt%磷酸水溶液(17:83)为流动相;检测波长为316nm;柱温35℃。理论板数按阿魏酸峰计算应不低于5000。
[0192]
具有有效成分含量测定结果见表12。
[0193]
表12酒炒不同时间所得当归饮片中阿魏酸的含量一览表
[0194]
试验号炒制时间(min)阿魏酸含量(%)130.0872250.1031370.1003
[0195]
由表12可以看出不同酒炒时间,所得当归饮片中阿魏酸的数据之间p<0.05,差别具有统计学意义,其中酒炒5min时,阿魏酸含量最高,说明本发明实施例1中的酒炒时间有利于最大程度的保留阿魏酸,防止阿魏酸分解。
[0196]
七)温经汤制备的对照实验
[0197]
本实验例中温经汤制备的对照实验均采用实施例1中炮制的甘草gc1、川芎cx1、芍药sy1、牡丹皮md1、人参rs1、牛膝nx1、当归dg1、莪术和桂心作为原料。
[0198]
71)是否加盖考察
[0199]
对比例1为实施例1中温经汤的制备的对比实验,不同之处仅在于,对比例1在煎煮过程中未加盖,所得温经汤冻干粉标记为d1。
[0200]
分别对温经汤冻干粉m1和d1中的易挥发成分桂皮醛和丹皮酚含量进行测定,具体结果如下:
[0201]
表13是否加盖对挥发性成分峰面积的影响
[0202][0203]
由表13可以看出,温经汤冻干粉m1中的桂皮醛含量明显高于温经汤冻干粉d1,因此,加盖煎煮和不加盖煎煮对于挥发性成分桂皮醛的影响较大,所以需要加盖煎煮。
[0204]
72)文火煎煮时间考察
[0205]
对比例2~4为实施例1中温经汤的制备的对比实验,不同之处仅在于:
[0206]
对比例2在煎煮过程中文火煎煮40min,此时剩余水量275ml;
[0207]
对比例3在煎煮过程中文火煎煮50min,此时剩余水量270ml;
[0208]
对比例4在煎煮过程中文火煎煮70min,此时剩余水量185ml;
[0209]
对比例2~4中剩余水量略多或略少,有效成分的溶出不稳定。
[0210]
73)过滤方式及目数的考察
[0211]
对比例5为实施例1中温经汤的制备的对比实验,不同之处仅在于:
[0212]
对比例5中采用8层纱布过滤,过滤用时为50s;
[0213]
针对对比例5和实施例1,统计出膏率,具体见下表:
[0214]
表14不同过滤方式的出膏率一览表
[0215]
滤过介质得率(%)8层纱布25.67100目筛网27.90
[0216]
由表14可以看出,采用不同过滤介质,出膏率差异不大。但是纱布过滤会吸附一定体积的煎煮液,损失较多,并且操作不方便,因此,从方便性以及实际生产可操作性的角度考虑,采用100目标准筛网作为滤过介质,在试验中进行固液分离。
[0217]
74)浓缩方式的考察
[0218]
对比例6~7为实施例1中温经汤的制备的对比实验,不同之处仅在于:
[0219]
对比例6中采用水浴加热浓缩后冻干,所得温经汤冻干粉标记为d6;
[0220]
对比例7中采用旋蒸加热浓缩后冻干,所得温经汤冻干粉标记为d7;
[0221]
分别对实施例1和对比例6~7中的温经汤冻干粉m1、d6~d7复溶后所得药液,采用超高效液相色谱法进行有效成分含量测定,具体结果见表15~16及图10~13。
[0222]
表15温经汤冻干粉m1和d6的挥发性成分峰面积一览表
[0223][0224]
表16温经汤冻干粉m1和d7的挥发性成分峰面积一览表
[0225][0226][0227]
表15~16及图10~13中可以看出采用加热浓缩的方式都将导致挥发性物质桂皮醛和丹皮酚的极大损失,而不加热浓缩不会导致挥发性物质损失,有助于最大程度的保留
温经汤的有效成分。
[0228]
显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。