1.本发明涉及一种适合工业化生产的索拉菲尼关键中间体4-氯吡啶-2-甲酸甲酯的方法,属于有机合成领域。
背景技术:
2.nature chemistry, 2022, vol. 14, # 1, p. 78
ꢀ–ꢀ
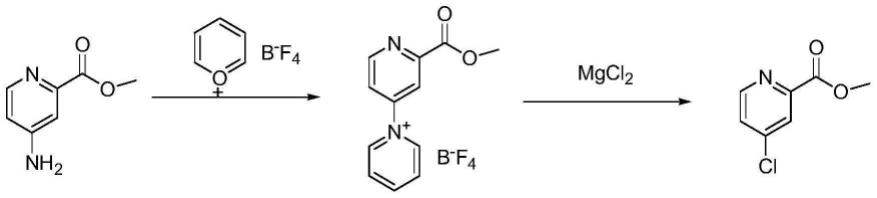
84公开了中间体4-氯吡啶-2-甲酸甲酯的制备方法,其路径如图1所示。该方法以4-氨基吡啶-2-甲酸甲酯为起始原料,再用吡喃鎓四氟硼酸盐与氨基形成杂环后,再用氯化镁提供氯源进行氯代。其中吡喃鎓四氟硼酸盐用量为1.5eq,其价格较贵且较难获得。同样4-氨基吡啶-2-甲酸甲酯价格较贵,所以该路线适合研究,不适合大规模生产。
技术实现要素:
3.本发明目的是为了开发出一种操作简单,生产成本低,适合工业化生产4-氯吡啶-2-甲酸甲酯的工艺。
4.为实现上述目的,本发明提供如下技术方案:一种适合工业化生产的索拉菲尼关键中间体4-氯吡啶-2-甲酸甲酯的方法,所述方法包括以下步骤:(1)2-吡啶甲酸与氯代试剂在催化剂作用下反应生成4-氯吡啶-2-甲酰氯盐酸盐;(2)将反应生成的4-氯吡啶-2-甲酰氯盐酸盐溶液减压脱溶;(3)添加溶剂溶解酰氯;(4)添加甲醇,对4-氯吡啶-2-甲酰氯盐酸盐进行酯化反应,生成4-氯吡啶-2-甲酸甲酯盐酸盐粗品;(5)对4-氯吡啶-2-甲酸甲酯盐酸盐粗品减压脱溶;(6)将4-氯吡啶-2-甲酸甲酯盐酸盐粗品加入水中,制成水溶液;(7)使用活性炭对水溶液进行脱色除焦;(8)脱色除焦后的水溶液进行游离得到4-氯吡啶-2-甲酸甲酯粗品;(9)使用二氯甲烷萃取水浮液,获得有机层;(10)对有机层干燥浓缩得4-氯吡啶-2-甲酸甲酯粗品;(11)将4-氯吡啶-2-甲酸甲酯粗品重结晶,制得高纯度4-氯吡啶-2-甲酸甲酯。
5.进一步地,所述催化剂是氯化钠、溴化钠或碘化钠中的一种。
6.进一步地,所述氯代试剂是草酰氯、氯化亚砜或三氯氧磷的一种。
7.进一步地,所述氯代试剂的当量为:2.5-8.0。
8.进一步地,所述催化剂的当量为:0.1-2.0。
9.进一步地,所述2-吡啶甲酸与氯代试剂在60-110℃下反应生成4-氯吡啶-2-甲酰氯。
10.进一步地,所述酯化反应的温度为:0-40℃。
11.进一步地,所述对水悬浮液进行游离是指向水悬浮液中滴加碳酸氢钠、碳酸钠或氢氧化钠水溶液至ph=6-7。
12.进一步地,所述游离温度为:0-35℃。
13.进一步地,所述重结晶是指将所述4-氯吡啶-2-甲酸甲酯粗品的正庚烷和正庚烷、乙酸乙酯以及醇水体系溶液加热至50-55℃溶解,搅拌0.5h后降温至10-15℃析晶,抽滤,滤饼干燥制得4-氯吡啶-2-甲酸甲酯纯品。
14.与现有技术相比,本发明的有益效果在于:该反应的机理如图2所示。
15.本发明采用成本较低的2-吡啶甲酸做原料,对氯代试剂,反应的投料比,氯代和酯化温度,催化剂种类及用量,酯化溶剂进行了改进,提高了转化率。反应结束,处理得到4-氯吡啶-2-甲酸甲酯盐酸盐粗品。
16.区别于其他工艺,我们通过对4-氯吡啶-2-甲酸甲酯盐酸盐粗品用热水溶解,用活性炭进行脱色除焦。除焦后再进行游离得到4-氯吡啶-2-甲酸甲酯粗品。将焦油在游离前除去,避免了焦油带到重结晶步骤,影响产品的析晶。在该步中对活性炭用量,调碱游离时碱的种类及其当量,调碱温度进行优化,提高了4-氯吡啶-2-甲酸甲酯盐酸盐粗品的质量。我们通过对重结晶体系进行改进,可提高纯度及收率。
17.根据反应机理,氯化亚砜在其中既做酰化试剂,又做氯代试剂。理论用量为2个当量。本发明设计当量为:2.5-8.0。氯代试剂优选草酰氯、氯化亚砜或三氯氧磷。溴化钠在其中可能与氯化亚砜形成溴化亚砜,更容易与原料反应。
18.上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。
附图说明
19.图1为背景技术部分介绍的反应路径;图2为本发明一实施例所示的反应路径。
具体实施方式
20.下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
21.实施例1:1000l的四口瓶中加入100.0g 2-吡啶甲酸,83.62g溴化钠,410ml氯化亚砜。控温80-85℃反应6h取样(取样加甲醇衍生化)监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。减压脱溶后加400ml甲苯溶解酰氯,降温至30-40℃,向其中滴加78.78g无水甲醇进行酯化。在此温度下反应1h取样监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。
22.后处理:减压脱溶,得到4-氯吡啶-2-甲酸甲酯盐酸盐粗品。将该粗品用1760ml的水悬浮,加热至50-55℃。在此温度下保温0.5h,向其中加入5.0g活性炭,搅拌0.5h。趁热过滤,滤饼加100.0ml热水润洗,抽滤。合并滤液,滤液降温至室温,控温10-15℃向其中滴加饱和na2co3水溶液至ph=6-7。向其中加入300ml*2二氯甲烷萃取两遍。静置分层,合并有机层。有机层干燥浓缩得111.5g 4-氯吡啶-2-甲酸甲酯粗品。将该粗品用890.0ml正庚烷进行重
结晶。加热至50-55℃溶解。在此温度下搅拌0.5h。降温至10-15℃析晶。保温0.5h后抽滤。滤饼真空干燥得78g 4-氯吡啶-2-甲酸甲酯纯品。收率56%,纯度99.2%实施例2:1000l的四口瓶中加入100.0g 2-吡啶甲酸,12.58g碘化钠,477ml草酰氯。控温60-65℃反应9h取样(取样加甲醇衍生化)监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。减压脱溶后加400ml二氯甲烷溶解酰氯,降温至20-30℃,向其中滴加105.04g无水甲醇进行酯化。在此温度下反应1h取样监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。
23.后处理:减压脱溶,得到4-氯吡啶-2-甲酸甲酯盐酸盐粗品。将该粗品用1800ml的水悬浮,加热至50-55℃。在此温度下保温0.5h,向其中加入5.0g活性炭,搅拌0.5h。趁热过滤,滤饼加热水润洗,抽滤。合并滤液,滤液降温至室温,控温0-5℃向其中滴加液碱naoh水溶液至ph=6-7。向其中加入300ml*2二氯甲烷萃取两遍。静置分层,合并有机层。有机层干燥浓缩得120.0g 4-氯吡啶-2-甲酸甲酯粗品。将该粗品用960.0ml正庚烷进行重结晶。加热至50-55℃溶解。在此温度下搅拌0.5h。降温至10-15℃析晶。保温0.5h后抽滤。滤饼真空干燥得82.0g 4-氯吡啶-2-甲酸甲酯纯品。收率58.8%,纯度99.0%实施例3:1000l的四口瓶中加入100.0g 2-吡啶甲酸,47.89g氯化钠,526.2ml三氯氧磷。控温100-105℃反应3h取样(取样加甲醇衍生化)监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。减压脱溶后,降温至10-20℃向其中滴加500ml无水甲醇进行酯化。在此温度下反应1h取样监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。
24.后处理:减压脱溶,得到4-氯吡啶-2-甲酸甲酯盐酸盐粗品。将该粗品用1800ml的水悬浮,加热至50-55℃。在此温度下保温0.5h,向其中加入10.0g活性炭,搅拌0.5h。趁热过滤,滤饼加热水润洗,抽滤。合并滤液,滤液降温至室温,控温30-35℃向其中滴加饱和nahco3水溶液至ph=6-7。向其中加入300ml*2二氯甲烷萃取两遍。静置分层,合并有机层。有机层干燥浓缩得105.0g 4-氯吡啶-2-甲酸甲酯粗品。将该粗品用840.0ml正庚烷进行重结晶。加热至50-55℃溶解。在此温度下搅拌0.5h。降温至10-15℃析晶。保温0.5h后抽滤。滤饼真空干燥得82.0g 4-氯吡啶-2-甲酸甲酯纯品。收率52.4%,纯度99.1%实施例4:1000l的四口瓶中加入100.0g 2-吡啶甲酸,62.5g碘化钠,200ml氯化亚砜。控温80-85℃反应5h取样(取样加甲醇衍生化)监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。减压脱溶后加400ml二氯甲烷溶解酰氯,降温至0-10℃,向其中滴加52.52g无水甲醇进行酯化。在此温度下反应1h取样监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。
25.后处理:减压脱溶,得到4-氯吡啶-2-甲酸甲酯盐酸盐粗品。将该粗品用1000ml的水悬浮,加热至50-55℃。在此温度下保温0.5h,向其中加入10.0g活性炭,搅拌0.5h。趁热过滤,滤饼加热水润洗,抽滤。合并滤液,滤液降温至室温,控温20-25℃向其中滴加饱和na2co3水溶液至ph=6-7。向其中加入300ml*2二氯甲烷萃取两遍。静置分层,合并有机层。有机层干燥浓缩得125.0g 4-氯吡啶-2-甲酸甲酯粗品。将该粗品用125.0ml乙酸乙酯加热至40-45℃溶解。在此温度下滴加1250.0ml正庚烷,保温搅拌0.5h。降温至10-15℃析晶。保温0.5h后抽滤。滤饼真空干燥得80.0g 4-氯吡啶-2-甲酸甲酯纯品。收率57.44%,纯度99.6%实施例5:1000l的四口瓶中加入100.0g 2-吡啶甲酸,62.5g碘化钠,200ml氯化亚
砜。控温80-85℃反应5h取样(取样加甲醇衍生化)监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。减压脱溶后加400ml二氯甲烷溶解酰氯,降温至10-15℃,向其中滴加52.52g无水甲醇进行酯化。在此温度下反应1h取样监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。
26.后处理:减压脱溶,得到4-氯吡啶-2-甲酸甲酯盐酸盐粗品。将该粗品用1000ml的水悬浮,加热至50-55℃。在此温度下保温0.5h,向其中加入10.0g活性炭,搅拌0.5h。趁热过滤,滤饼加热水润洗,抽滤。合并滤液,滤液降温至室温,控温20-25℃向其中滴加饱和na2co3水溶液至ph=6-7。向其中加入300ml*2二氯甲烷萃取两遍。静置分层,合并有机层。有机层干燥浓缩得122.0g 4-氯吡啶-2-甲酸甲酯粗品。将该粗品用1830.0ml水进行重结晶。加热至55-60℃溶解。在此温度下搅拌0.5h。降温至10-15℃析晶。保温0.5h后抽滤。滤饼真空干燥得70.5g 4-氯吡啶-2-甲酸甲酯纯品。收率50.6%,纯度99.7%实施例6:1000l的四口瓶中加入100.0g 2-吡啶甲酸,62.5g碘化钠,200ml氯化亚砜。控温80-85℃反应5h取样(取样加甲醇衍生化)监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。减压脱溶后加400ml二氯甲烷溶解酰氯,降温至10-15℃,向其中滴加52.52g无水甲醇进行酯化。在此温度下反应1h取样监控至原料2-吡啶甲酸耗至1.0%以下,主产4-氯吡啶-2-甲酸甲酯>90.0%后,停止反应。
27.后处理:减压脱溶,得到4-氯吡啶-2-甲酸甲酯盐酸盐粗品。将该粗品用1000ml的水悬浮,加热至50-55℃。在此温度下保温0.5h,向其中加入10.0g活性炭,搅拌0.5h。趁热过滤,滤饼加热水润洗,抽滤。合并滤液,滤液降温至室温,控温20-25℃向其中滴加饱和na2co3水溶液至ph=6-7。向其中加入300ml*2二氯甲烷萃取两遍。静置分层,合并有机层。有机层干燥浓缩得124.0g 4-氯吡啶-2-甲酸甲酯粗品。将该粗品用124.0ml乙醇加热至30-35℃溶解。在此温度下滴加620.0ml水,保温搅拌0.5h。降温至10-15℃析晶。保温0.5h后抽滤。滤饼真空干燥得75.0g 4-氯吡啶-2-甲酸甲酯纯品。收率53.85%,纯度99.7%以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。