1.本发明涉及乙酰化胞嘧啶三磷酸盐及其中间体的制备方法。
背景技术:
2.随着核酸药物的发展,mrna被视为了一种可以用于药物制造的新选择。1990年,一段mrna被注射进入小鼠体内,并成功编码出了蛋白质。这段mrna则是通过一种名为体外转录的技术得到的。随后,一项1992年的研究发现注射抗利尿激素编码mrna可以成功诱导大鼠的下丘脑的神经活动。虽然mrna显示出很好的生物活性,但是受限于其自身的不稳定性,强免疫原性和体内递送困难,mrna远远无法应用于临床疾病治疗中。
3.修饰核苷酸的加入,可以降低mrna的自身免疫原性,提高mrna自身稳定性,进一步增强mrna在目标细胞内的表达时间和表达效率,使mrma可以真正用于制药领域。其中moderna和biontech分别采用1-甲基假尿苷(cn110511939a,cn104114572a,cn103974724a)和假尿苷(us10232055b2,us9597380b2,us9163213b2),极大的降低了mrna的免疫原性,并且提高了mrna在靶细胞内表达目的蛋白的时间和总量,为mrna制成covid-19疫苗并成功上市奠定了基础。但是假尿苷及其衍生物合成极为困难,价格昂贵,并且其专利都被moderna,university of pennsylvania,biontech等寡头垄断。
4.以下列出了现有技术中关于n4乙酰基胞苷的常用合成方式,发明人对其中的部分方法进行了尝试,发现下述反应具有反应条件比较剧烈,反应时间长,副反应很多,纯化困难等缺点,完全无法得到原文献或专利中报道的产率。
[0005][0006]
综上所述,目前面临的主要问题时现阶段市售的乙酰化胞嘧啶三磷酸盐价格极为昂贵,且大量合成难度大,副反应多,最终产物极性大。
技术实现要素:
[0007]
为了解决现有技术中乙酰化胞嘧啶三磷酸价格昂贵,且大量合成难度大,副反应多,最终产物极性大等问题,本发明提供一种乙酰化胞嘧啶三磷酸盐及其中间体的制备方
法。本发明的合成方法极大缩短了反应时间、节约成本、简化反应条件,且反应产物较为单一。
[0008]
本发明第一方面提供一种如式3所述的化合物的制备方法,其包括以下步骤:将化合物2进行脱乙酰基反应,得如式3所示的化合物;
[0009][0010]
较佳地,所述脱乙酰基反应为所述化合物2与氨甲醇反应。
[0011]
所述氨甲醇与所述化合物2的摩尔比较佳地为(2-12):1,优选(10-12):1;例如10.8:1、11.66:1、8.3:1、5:1、3.3:1。
[0012]
所述氨甲醇的浓度较佳地为1-7mol/l,例如3mol/l、5mol/l。
[0013]
所述乙酰基反应的反应温度较佳地为0℃-10℃,例如5℃。
[0014]
所述乙酰基反应的反应时间较佳地为4min-10min,例如5min。
[0015]
所述乙酰基反应优选地还包括以下后处理步骤:在粗产品中加入乙醚溶液收集沉淀,较佳地,所述沉淀使用乙醇重结晶或使用柱层析进行纯化。
[0016]
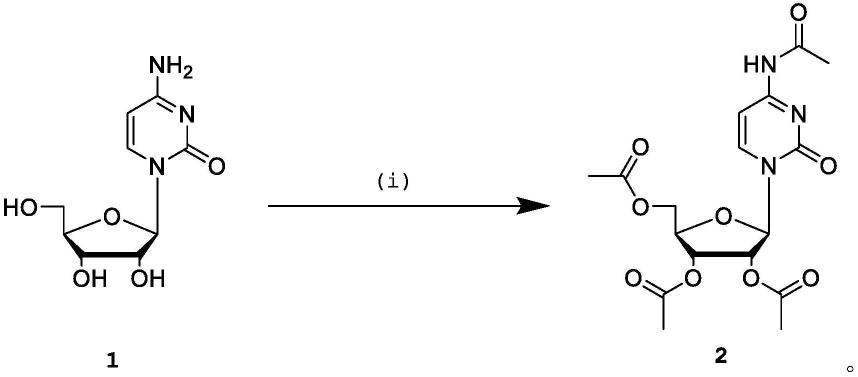
在某一较佳实施方案中,通过将化合物1进行乙酰基保护反应得所述化合物2。
[0017]
优选地,所述化合物2的制备方法包括以下步骤:在极性非质子溶剂中,将所述化合物1与乙酰化试剂进行乙酰基保护反应,得所述化合物2。
[0018]
所述极性非质子溶剂较佳地为n,n-二甲基甲酰胺(dmf)、乙腈、四氢呋喃或吡啶。
[0019]
所述乙酰化试剂较佳地为醋酸酐或乙酰氯。
[0020]
所述乙酰化试剂与所述化合物1的摩尔比较佳地为(3-7):1,例如3:1、4:1,更佳地为(5-7):1,例如5:1、6:1、7:1。
[0021]
所述极性非质子溶剂与所述化合物1的体积质量比较佳地为10-20ml/g,例如16.7ml/g、20ml/g、10ml/g。
[0022]
所述乙酰基保护反应的反应温度较佳地为10℃-25℃,例如15℃。
[0023]
所述乙酰基保护反应的反应时间较佳地为5min-15min,例如15min、10min、5min。
[0024]
所述乙酰基保护反应较佳地还包括以下后处理步骤:反应结束后,加入碱性溶液调节至中性,再加入有机溶剂进行萃取,然后有机相干燥浓缩即可。
[0025]
所述有机溶剂可为本领域常规,例如二氯甲烷、三氯甲烷或乙酸乙酯。
[0026]
所述碱性溶液较佳地为饱和碳酸氢钠水溶液。
[0027]
本发明第二方面提供一种化合物6的制备方法,其通过以下步骤制备:
[0028]
(1)将本发明第一方面所述的如式3所示的化合物制备为化合物4;
[0029]
(2)将化合物4制备为化合物5;
[0030]
(3)将化合物5制备为化合物6;
[0031][0032][0033]
本发明第二方面的制备方法较佳地为在丙酮溶液的存在下,将高氯酸钠与化合物5进行离子交换反应,得化合物6。
[0034]
较佳地将(bu3n)2h4p2o7、bu3n与化合物4进行磷脂化反应,并加入水或三乙胺碳酸盐(teab)缓冲液进行淬灭反应,得化合物5。
[0035]
较佳地在三磷酸甲酯中,将所述如式3所示的化合物与三氯氧磷进行磷酰化反应,得化合物4。
[0036]
所述磷脂化反应中,所述(bu3n)2h4p2o7与化合物4的摩尔比较佳地为(5-10):1,例如6:1、8:1、10:1。
[0037]
所述磷脂化反应中,所述bu3n与化合物4的摩尔比为较佳地(4-10):1;例如6:1、5.88:1、5:1、7:1。
[0038]
所述磷脂化反应中,所述水或teab与所述化合物4的摩尔比较佳地为(1-6):1,例如5.4:1、1.41:1、1.63:1、4.62:1。
[0039]
所述磷脂化反应中,反应温度较佳地为5℃-20℃,例如为15℃、10℃。
[0040]
所述磷脂化反应中,反应时间较佳地为5min-20min,例如为10min、15min。
[0041]
在某一较佳实施方案中,进行所述淬灭反应后的反应液用二氯甲烷进行萃取,并将水相进行冻干浓缩即可得到化合物5粗产品。
[0042]
在某一较佳实施方案中,将得到的所述化合物5粗产品使用deae sepharose a250离子交换树脂吸附,并用teab缓冲液进行洗脱,对洗脱组分进行浓缩干燥得到纯化的化合物5。
[0043]
所述磷酰化反应中,所述三氯氧磷与如式3所示的化合物的摩尔比较佳地为(1-2):1,例如1.5:1。
[0044]
所述磷酰化反应,所述三磷酸甲酯与所述如式3所示的化合物的体积质量比较佳地为10-20ml/g,例如14.9ml/g、12ml/g、17ml/g。
[0045]
所述磷酰化反应,反应温度较佳地为-10℃-20℃,例如为0℃、5℃、10℃、15℃。
[0046]
所述磷酰化反应,反应时间较佳地为20min-40min,例如为30min。
[0047]
所述离子交换反应中,所述高氯酸钠与化合物5的质量比较佳地为(3-17):1,例如15:1、9.67、5.1、7.73、16.1:1、3.8:1。
[0048]
所述离子交换反应中,所述丙酮溶液与所述化合物5的体积质量比较佳地为10-32ml/g,例如30ml/g、15.6ml/g、26ml/g、10.4ml/g、31.25ml/g。
[0049]
所述离子交换反应中,反应时间较佳地为20min-60min,例如为30min、40min。
[0050]
在某一较佳实施方案中,进行所述离子交换反应后包括将获得的悬浊体进行过滤、洗涤和干燥的步骤。
[0051]
本发明的积极进步效果在于:
[0052]
提供一种乙酰化胞嘧啶三磷酸盐及其中间体的制备方法。本发明的合成方法极大缩短了反应时间(反应过程小于2小时)、节约成本、简化反应条件,且反应产物较为单一。此外,经过体外转录和细胞活性验证后可知本方法合成的乙酰化胞嘧啶三磷酸拥有很好的活性与生物利用度。
附图说明
[0053]
图1为不同比例的4氨基乙酰化胞嘧啶替换胞嘧啶后mrna的电泳检测示意图。
[0054]
图2为使用电泳生物分析仪检测egfp mrna的结果示意图。
[0055]
图3为pbmc细胞的阳性率检测示意图。
[0056]
图4为egfp mrna在t细胞中的蛋白表达效率示意图。
[0057]
图5为乙酰化胞嘧啶三磷酸钠盐的hplc谱图。
具体实施方式
[0058]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0059]
实施例1:4-乙酰基-胞嘧啶核苷三磷酸钠盐的快速全合成:
[0060][0061]
其中(i)ac2o/无水dmf;(ii)nh3,7m in ch3oh;(iii)pocl3/po(ome)3;(iv)bu3n,(bu3n)2h4p2o7/mecn;(v)naclo4/丙酮。
[0062]
(i)采用不同的原料用量比分别制备化合物2。
[0063]
①
:胞嘧啶核苷1(15.0g,61.7mmol,1eq)25℃溶解于dmf(250.0ml)缓慢滴加过量醋酸酐(29.35ml,308.5mmol,5eq)/乙酰氯(21.94ml,308.5mmol,5eq)。5分钟后反应体系内缓慢加入饱和碳酸氢钠水溶液至中性。反应液用二氯甲烷(250ml
×
3)萃取,将有机相进行旋转浓缩干燥后得到化合物2为白色粉末(24.8g,60.5mmol,98%收率)。
[0064]
②
:胞嘧啶核苷1(15.0g,61.7mmol,1eq)25℃溶解于dmf(250.0ml)缓慢滴加过量醋酸酐(23.48ml,246.8mmol,4eq)/乙酰氯(17.55ml,246.8mmol,4eq)。5分钟后反应体系内缓慢加入饱和碳酸氢钠水溶液至中性。反应液用二氯甲烷(250ml
×
3)萃取,将有机相进行旋转浓缩干燥后得到化合物2为白色粉末(14.6g,35.8mmol,59%收率)。
[0065]
③
:胞嘧啶核苷1(15.0g,61.7mmol,1eq)15℃溶解于dmf(250.0ml)缓慢滴加过量醋酸酐(35.22ml,370.2mmol,6eq)/乙酰氯(26.32ml,370.2mmol,6eq)。15分钟后反应体系内缓慢加入饱和碳酸氢钠水溶液至中性。反应液用二氯甲烷(250ml
×
3)萃取,将有机相进行旋转浓缩干燥后得到化合物2为白色粉末(24.6g,60.3mmol,98%收率)。
[0066]
④
:胞嘧啶核苷1(15.0g,61.7mmol,1eq)10℃溶解于dmf(300.0ml)缓慢滴加过量醋酸酐(41.09ml,431.9mmol,7eq)/乙酰氯(37.43ml,431.9mmol,7eq)。10分钟后反应体系内缓慢加入饱和碳酸氢钠水溶液至中性。反应液用二氯甲烷(250ml
×
3)萃取,将有机相进行旋转浓缩干燥后得到化合物2为白色粉末(24.6g,60.3mmol,98%收率)。
[0067]
⑤
:胞嘧啶核苷1(15.0g,61.7mmol,1eq)25℃溶解于dmf(150.0ml)缓慢滴加等量醋酸酐(17.61ml,185.1mmol,3eq)/乙酰氯(13.16ml,185.1mmol,3eq)。5分钟后反应体系内缓慢加入饱和碳酸氢钠水溶液至中性。反应液用二氯甲烷(250ml
×
3)萃取,将有机相进行旋转浓缩干燥后得到化合物2为白色粉末(43.8g,33.8mmol,55%收率)。
[0068]
⑥
:胞嘧啶核苷1(15.0g,61.7mmol,1eq)15℃溶解于dmf(150.0ml)缓慢滴加等量醋酸酐(17.61ml,185.1mmol,3eq)/乙酰氯(13.16ml,185.1mmol,3eq)。15分钟后反应体系内缓慢加入饱和碳酸氢钠水溶液至中性。反应液用二氯甲烷(250ml
×
3)萃取,将有机相进行旋转浓缩干燥后得到化合物2为白色粉末(43.8g,33.6mmol,54%收率)。
[0069]
对于上述六种配比关系制备得到的化合物2进行结构确定:
[0070]
(2r,3r,4r,5r)-5-(4-acetamido-2-oxopyrimidin-1(2h)-yl)-2-[0071]
(acetoxymethyl)-4-(prop-1-en-2-yloxy)tetrahydrofuran-3-yl acetate((2r,3r,4r,5r)-5-(4-乙酰氨基-2-羰基嘧啶-1(2h)-基)-2-(乙酰氧基甲基)-4-(丙-1-烯-2-氧基)四氢呋喃-3-基乙酸酯).1h-nmr(400mhz,cdcl3)δ8.74
–
8.65(m,1h),7.95(d,j=7.6hz,1h),7.53(d,j=7.6hz,1h),6.12(d,j=4.0hz,1h),5.55
–
5.43(m,1h),5.43
–
5.32(m,1h),4.48
–
4.44(m,1h),4.44
–
4.42(m,2h),2.31(s,3h),2.19(s,3h),2.15(s,3h),2.13(s,3h).esi-tof-ms m/z calcd for c
18h24
n3o
8
[m h]
410.2:found.410.2。
[0072]
在化合物1制备化合物2的乙酰基保护反应中,将饱和碳酸氢钠水溶液替换为饱和碳酸钠和稀氢氧化钠,结果发现反应产率会极大降低,根据tlc实验结果反馈反应产率会降至20%以下,因为会产生复杂的降解反应所以未对目标物进行相应的纯化工作,未得到具体数值。
[0073]
(ii)控制不同浓度的氨甲醇溶液和反应时间分别制备化合物3。
[0074]
a:化合物2(24.7g,60.0mmol)溶解于0℃的7m的氨甲醇溶液(100ml)反应5分钟,旋转蒸发干燥后取得透明油状物粗产品3。粗产品中加入0℃的乙醚溶液(400ml)后收集沉淀。沉淀可用50%的乙醇溶液进行重结晶取得化合物3(16.4g,57.5mmol,95%收率)为白色粉末。粗产品使用柱层析法也可取得很好的纯化分离效果。
[0075]
b:化合物2(24.7g,60.0mmol)溶解于5℃的5m的氨甲醇溶液(100ml)反应10分钟,旋转蒸发干燥后取得透明油状物粗产品3。粗产品中加入0℃的乙醚溶液(400ml)后收集沉淀。沉淀可用50%的乙醇溶液进行重结晶取得化合物3(16.4g,57.5mmol,95%收率)为白色粉末。粗产品使用柱层析法也可取得很好的纯化分离效果。
[0076]
c:化合物2(24.7g,60.0mmol)溶解于10℃的3m的氨甲醇溶液(100ml)反应10分钟,旋转蒸发干燥后取得透明油状物粗产品3。粗产品中加入0℃的乙醚溶液(400ml)后收集沉淀。沉淀可用50%的乙醇溶液进行重结晶取得化合物3(16.4g,57.5mmol,95%收率)为白色粉末。粗产品使用柱层析法也可取得很好的纯化分离效果。
[0077]
d:化合物2(24.7g,60.0mmol)溶解于10℃的1m的氨甲醇溶液(200ml)反应10分钟,旋转蒸发干燥后取得透明油状物粗产品3。粗产品中加入0℃的乙醚溶液(400ml)后收集沉淀。沉淀可用50%的乙醇溶液进行重结晶取得化合物3(15.5g,54.37mmol,90%收率)为白色粉末。粗产品使用柱层析法也可取得很好的纯化分离效果。
[0078]
e:化合物2(24.7g,60.0mmol)溶解于0℃的7m的氨甲醇溶液(200ml)反应13分钟,旋转蒸发干燥后取得透明油状物粗产品3。粗产品中加入0℃的乙醚溶液(400ml)后收集沉淀。沉淀可用50%的乙醇溶液进行重结晶取得化合物3(9.0g,31.4mmol,52%收率)为白色粉末。
[0079]
f:化合物2(24.7g,60.0mmol)溶解于0℃的7m的氨甲醇溶液(100ml)反应4分钟,旋转蒸发干燥后取得透明油状物粗产品3。粗产品中加入0℃的乙醚溶液(400ml)后收集沉淀。沉淀可用50%的乙醇溶液进行重结晶取得化合物3(16.4g,57.5mmol,96%收率)为白色粉末。粗产品使用柱层析法也可取得很好的纯化分离效果。
[0080]
g:化合物2(24.7g,60.0mmol)溶解于0℃的7m的氨甲醇溶液(100ml)反应3分钟,旋转蒸发干燥后取得透明油状物粗产品3。粗产品中加入0℃的乙醚溶液(400ml)后收集沉淀。沉淀可用50%的乙醇溶液进行重结晶取得化合物3(11.4g,39.87mmol,66%收率)为白色粉
末。
[0081]
化合物3进行结构确定:
[0082]
n-(1-((2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)acetamide(n-(1-((2r,3r,4s,5r)-3,4-二羟基-5-(羟甲基)四氢呋喃-2-基)-2-羰基-1,2-二氢嘧啶-4-基)乙酰胺)。
[0083]1h-nmr(400mhz,dmso-d6)δ8.31(d,j=7.5hz,1h),7.31(d,j=7.5hz,1h),5.88(d,j=2.6hz,1h),4.39
–
4.27(m,1h),4.25
–
4.12(m,2h),3.96(d,1h),3.84(d,1h),2.22(s,3h).esi-tof-ms m/z calcd for c
11h16
n3o
6
[m h]
286.1:found.286.1。
[0084]
在化合物2制备化合物3的脱乙酰基反应中,当氨甲醇与化合物2的摩尔比小于2时,或反应温度低于0℃时,或反应时间小于4min容易导致脱保护不完全,出现2
’3’5’
位置不均一脱保护现象,出现大量脱保护不完全的中间产物。
[0085]
当氨甲醇与化合物2的摩尔比大于12,或反应温度大于10℃,或反应时间大于10min时容易出现过度脱保护,将4n位置乙酰基脱保护甚至导致核苷的碱基分解或者戊糖部分分解。
[0086]
(iii)化合物3(4.5g,18.4mmol,1eq)溶解于po(ome)3(67.0ml),滴加-10℃的pocl3(4.24g,27.6mmol,2.57ml,1.50eq)。反应继续15℃搅拌30分钟。lcms检测至原料消失,并全部转化为化合物4(4.5g,18.4mmol,1eq)(esi-tof-ms m/z calcd for c
11h14
cl2n3o
7
[m h]
402.0:found.402.0)。
[0087]
(iv)在(iii)获得的化合物4的基础上,进一步在不同原料比例和反应条件下制备化合物5:
[0088]
①
:加入-10℃的bu3n-pyrophosphate((bu3n)2h4p2o7,三丁基焦磷酸铵)(0.60m in mecn,154ml,5eq)和bu3n(20.1g,108mmol,25.8ml,5.88eq)。反应继续15℃搅拌15分钟至lcms检测化合物4完全消失。反应体系内缓慢加入1m teab(三乙胺碳酸盐)缓冲液(100ml)淬灭反应。
[0089]
②
:加入-10℃的bu3n-pyrophosphate(0.60m in mecn,132ml,4eq)和bu3n(27.2g,108mmol,25.8ml,5.88eq)。反应继续15℃搅拌10分钟lcms检测化合物4不能完全消失,反应失败。
[0090]
③
:加入-10℃的bu3n-pyrophosphate(0.60m in mecn,198ml,6eq)和bu3n(20.1g,147mmol,35ml,8eq)。反应继续20℃搅拌15分钟至lcms检测化合物4完全消失。反应体系内缓慢加入超纯水1.3ml(72.2mmol,4eq)淬灭反应。
[0091]
④
:加入-10℃的bu3n-pyrophosphate(0.60m in mecn,277ml,9eq)和bu3n(17.1g,92mmol,21.9ml,5eq)。反应继续10℃搅拌5分钟至lcms检测化合物4完全消失。反应体系内缓慢加入1m teab缓冲液(26ml)淬灭反应。
[0092]
⑤
:加入-10℃的bu3n-pyrophosphate(0.60m in mecn,154ml,5eq)和bu3n(34.1g,184mmol,25.8ml,10eq)。反应继续5℃搅拌20分钟至lcms检测化合物4完全消失。反应体系内缓慢加入1m teab缓冲液(30ml)淬灭反应。
[0093]
⑥
:加入-10℃的bu3n-pyrophosphate(0.60m in mecn,154ml,5eq)和bu3n(23.6g,129mmol,30.3ml,7eq)。反应继续5℃搅拌20分钟至lcms检测化合物4完全消失。反应体系内缓慢加入1m teab缓冲液(85ml)淬灭反应。
[0094]
反应液用二氯甲烷(250ml
×
3)萃取,将水相进行冻干浓缩。可得到淡黄色油状化合物5粗产品。将化合物5粗产品用deae sepharose a250离子交换树脂进行吸附后,用0-1m teab缓冲液进行洗脱,收集相应的产物洗脱峰。对洗脱组分进行旋转蒸发干燥后可得到化合物5(9.5g,粗品)为透明油状液体。
[0095]
化合物5进行结构确定:
[0096]
triethylamine((2r,3s,4r,5r)-5-(4-acetamido-2-oxopyrimidin-1(2h)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl triphosphate(三乙基胺((2r,3s,4r,5r)-5-(4-乙酰氨基-2-羰基嘧啶-1(2h)-基)-3,4-二羟基四氢呋喃-2-基)甲基三磷酸酯)。esi-tof-ms m/z calcd for c
11h17
n3o
15-[m-h]-524.0:found.524.0。
[0097]
(v)在不同条件下制备化合物6:
[0098]
①
:将化合物5(9.5g,粗品)溶解于5m高氯酸钠/丙酮溶液(250ml)中搅拌30分钟,对悬浊液体进行减压抽滤,滤饼用0℃丙酮溶液清洗后进行减压干燥,可得到化合物6(6.3g,10.3mmol,56%收率)为白色粉末。
[0099]
②
:将化合物5(9.5g,粗品)溶解于5m高氯酸钠/丙酮溶液(150ml)中搅拌20分钟,对悬浊液体进行减压抽滤,滤饼用0℃丙酮溶液清洗后进行减压干燥,可得到化合物6(6.2g,10.2mmol,56%收率)为白色粉末。
[0100]
③
:将化合物5(9.5g,粗品)溶解于3m高氯酸钠/丙酮溶液(100ml)中搅拌40分钟,对悬浊液体进行减压抽滤,滤饼用25℃丙酮溶液清洗后进行减压干燥,可得到化合物6(6.2g,10.2mmol,56%收率)为白色粉末。
[0101]
④
:将化合物5(9.5g,粗品)溶解于4m高氯酸钠/丙酮溶液(100ml)中搅拌60分钟,对悬浊液体进行减压抽滤,滤饼用0℃丙酮溶液清洗后进行减压干燥,可得到化合物6(6.2g,10.2mmol,56%收率)为白色粉末。
[0102]
⑤
:将化合物5(9.5g,粗品)溶解于2m高氯酸钠/丙酮溶液(300ml)中搅拌60分钟,对悬浊液体进行减压抽滤,滤饼用0℃丙酮溶液清洗后进行减压干燥,可得到化合物6(6.2g,10.2mmol,56%收率)为白色粉末。
[0103]
对化合物6进行结构确认:
[0104]
sodium((2r,3s,4r,5r)-5-(4-acetamido-2-oxopyrimidin-1(2h)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl triphosphate(((2r,3s,4r,5r)-5-(4-乙酰氨基-2-羰基嘧啶-1(2h)-基)-3,4-二羟基四氢呋喃-2-基)甲基三磷酸钠盐)。1h nmr(500mhz,deuterium oxide)δ8.35(d,j=7.6hz,1h),7.26(d,j=7.6hz,1h),5.90(d,j=3.0hz,1h),4.38
–
4.18(m,5h),2.16(s,3h).
31
p nmr(202mhz,deuterium oxide)δ-7.94
–‑
8.56(m,1p),-11.23(d,j=19.7hz,1p),-22.32(t,j=19.7hz,1p).esi-tof-ms m/z calcd for c
11h17
n3o
15-[m-h]-524.0:found.524.0。通过hplc(柱子:shim-pack gist,5μm c18-aq,4.6*250mm,流速:1ml/min,流动相:乙腈/水=10/90(v/v))测定其纯度为100%。结果见图5。
[0105]
效果实施例1:对制备的乙酰化胞嘧啶三磷酸钠盐进行体外转录活性测试。
[0106]
将化合物6配置成为100mm水溶液,即100mm 4acctp。并分别按照4acctp/ctp
×
100%=50%,25%,10%,1%,0.1%混合成为不同比例的100mm 4acctp/ctp溶液。
[0107]
八个样品分别加入100mm atp溶液,100mm gtp溶液,100mm utp溶液各2μl。不同样
品添加100mm 4acctp,50%,25%,10%,1%,0.1%100mm 4acctp/ctp,100mm ctp。复合酶(40u/μl rna酶抑制剂(上海翊圣生物科技有限公司,货号:10603es05),0.1u/μl无机焦磷酸酶,1000u/μl t7 rna聚合酶(上海翊圣生物科技有限公司,货号:10618es90),0.2m氯化镁)2μl。10
×
反应液(400mm tris-hcl,60mm mgcl2,20mm亚精胺,100mm dtt,ph 7.9)2μl。egfp dna模板(购自金维智)1μg补加超纯水至20μl。37℃孵育2小时后加入1μl dna酶,继续37℃孵育30分钟。反应体系经过6小时37℃孵育后,由1%琼脂糖凝胶进行电泳检测,结果见图1。
[0108]
对转录完成产物进行2.5m氯化锂沉淀或利用deae纤维素色谱柱纯化。纯化完成后将egfp mrna样品加热至65℃保持5min后迅速转至冰上冷却至0℃,加入10
×
加帽缓冲液,10mm gtp,2mm s-腺苷甲硫氨酸,10u/μl牛痘加帽酶37℃孵育30min。
[0109]
加帽完成后的egfp mrna利用deae纤维素色谱柱进行纯化。并利用labchip gxii touch ht(perkinelmer)进行egfp mrna纯度测试,结果见图2。纯化完成后的egfp mrna放入-80℃进行保存。
[0110]
效果实施例2:利用4-氨基乙酰化胞嘧啶修饰的mrna对靶细胞进行转染并检测靶细胞的阳性率和egfp mrna的表达效率。
[0111]
将lonza电转液按a/b=0.82/0.18比例混合,取10μg不同的egfp mrna样品与相应电转液混合均匀。
[0112]
将cd3/cd28抗体(cd3抗体:biolegend 317302,cd28抗体:biolegend302902)孵化,加入d-pbs缓冲液,调整浓度为5μg/ml,混匀后加入6孔板进行37℃孵育。
[0113]
将低温冻存的pbmc(外周血单个核细胞,购自妙顺生物科技有限公司)进行复苏后,1
×
107细胞分装至1.5ml离心管内,加入电转液及egfp mrna样本混合液,混合均匀后进行电击。电穿孔完成后将细胞悬液加入含2%fbs的aim-v培养液中37℃,5%co2培养。
[0114]
4-6小时后取出cd3/cd28抗体包被平板,去除d-pbs缓冲液,将电转完成的pbmc转移至抗体包被平板中并补加il-2(山东港泉药业)至500u/ml,37℃,5%co2培养。
[0115]
培养4-5天后将pbmc细胞转移出抗体包被平板后加入新鲜含2%fbs的aim-v培养基(gibco),并加入il-2抗体至浓度200u/ml。分别在电转后1天和14天收集细胞样本,并用流式细胞仪检测样本pbmc细胞的阳性率(如图3所示)和egfp mrna的表达效率(如图4所示)。
[0116]
经检测通过本方法合成制备的4氨基乙酰化胞嘧啶三磷酸钠盐替换胞嘧啶的egfp mrna都可以让pbmc细胞产生较好的阳性率,同时当替换比率达到50%时可以使egfp mrna拥有最好的表达效率。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
