治疗新冠肺炎口服药物s-217622的制备方法
技术领域
1.本发明属于有机合成路线设计及其原料药和中间体制备技术领域,特别涉及一种治疗新冠肺炎口服药物s-217622的制备方法。
背景技术:
2.s-217622是由日本盐野义公司开发的一种治疗新冠肺炎口服药物,具有高效抑制新冠病毒3cl酶并发挥抗病毒的作用。临床前试验表明,s-217622在体外对3cl酶活性作用较强,ic
50
值为0.013μm,ec
50
值为0.37μm。动物试验表明,s-217622在大鼠、猴子及狗中口服给药具有高吸收率和低清除率,其在猴子和狗中的半衰期分别约为10和30小时。同时,体外试验还证实s-217622对多种新冠病毒变异株均具有较强的抗病毒活性,针对奥密克戎变异株的ec
50
值约为0.29μm,未发生明显减低。目前,该药已经完成iib期临床试验,基于其临床试验结果,盐野义公司已经向日本厚生劳动省申请生产及上市许可。
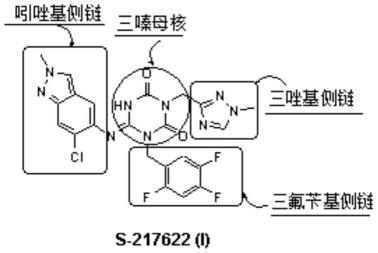
3.s-217622(i)的化学名为:(6e)-6-[(6-氯-2-甲基-2h-吲唑-5-基)亚氨基]二氢-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮,其结构式如下:
[0004][0005]
分析s-217622的分子结构组成可以发现,该化合物是由“三嗪”母核和“吲唑基”、“三唑基”以及“三氟苄基”三个“侧链”所构成。
[0006][0007]
基于上述分析,借鉴“逆向合成法”思路,盐野义公司的研究文献“discovery of s-217622,a non-covalent oral sars-cov-2 3cl protease inhibitor clinical candidate for treating covid-19”(biorxiv,posted january 26,2022)公开了一种s-217622的合成路线:
1h-1,2,4-三唑(iii)和n,n
’‑
羰基二咪唑在碱促进剂作用下发生环合反应生成6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(iv);所述6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(iv)与2,4,5-三氟苄基溴在缚酸剂作用下发生取代反应得到6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(v),所述6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(v)与6-氯-2-甲基-2h-吲唑-5-胺发生缩合反应生成(6e)-6-[(6-氯-2-甲基-2h-吲唑-5-基)亚氨基]二氢-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(s-217622,i)。
[0016]
反应路线示意如下:
[0017][0018]
此外,本发明还提供如下附属技术方案:
[0019]
所述环合反应的原料2-乙基-2-异硫脲氢溴酸盐(ii)、1-甲基-3-异氰酸酯甲基-1h-1,2,4-三唑(iii)和n,n
’‑
羰基二咪唑的投料摩尔比为1:1~1.5:1~2,优选1:1.25:1.5。
[0020]
所述环合反应的碱促进剂为三乙胺、吡啶、2,6-二甲基吡啶、4-二甲氨基吡啶、n-甲基吗啉、n-乙基吗啉、二异丙基乙胺、1,5-二氮杂二环[4.3.0]-壬-5-烯、1,8-二氮杂双环[5.4.0]-十一-7-烯或1,4-二氮杂二环[2.2.2]辛烷,优选1,8-二氮杂双环[5.4.0]-十一-7-烯或1,5-二氮杂二环[4.3.0]-壬-5-烯或1,4-二氮杂二环[2.2.2]辛烷。
[0021]
所述环合反应的碱促进剂与原料2-乙基-2-异硫脲氢溴酸盐(ii)的投料摩尔比为1~1.5:1,优选1.2~1.3:1。
[0022]
所述环合反应的溶剂为四氢呋喃、乙腈、二氧六环、二甲亚砜、n,n-二甲基甲酰胺或乙腈,优选n,n-二甲基甲酰胺或乙腈。
[0023]
所述环合反应的温度为-10~25℃,优选0~5℃。
[0024]
所述取代反应原料6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1,3,5-三嗪-2,4(1h,3h)
‑ꢀ
二酮(iv)与2,4,5-三氟苄基溴的投料摩尔比为1:1~2,优选1:1.5。
[0025]
所述取代反应的缚酸剂为氢化钠、氢化钾、氢氧化钠、氢氧化钾、叔丁醇钠、叔丁醇钾、甲醇钠、乙醇钠、碳酸钠、碳酸钾或碳酸铯,优选碳酸钾或碳酸铯。
[0026]
所述取代反应的溶剂为甲苯、二甲苯、二氧六环、二甲亚砜、n,n-二甲基甲酰胺或乙腈,优选乙腈。
[0027]
所述取代反应的温度为50~100℃,优选75~85℃。
[0028]
所述缩合反应原料6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(v)和6-氯-2-甲基-2h-吲唑-5-胺的投料摩尔比为1:1~2,优选1:1.5。
[0029]
所述缩合反应的溶剂为四氢呋喃、乙腈、二氧六环、异丙醇、正丁醇或叔丁醇,优选叔丁醇。
[0030]
所述缩合反应的温度为50~120℃,优选80~90℃。
[0031]
有益效果
[0032]
本发明所涉及的抗新冠药物s-217622的制备方法,通过已知原料,依次经过常见单元反应如环合反应、取代反应和缩合反应等,使其制备过程更加简洁,条件温和且安全环保。尤其是通过一步环合反应,减少了保护及脱保护步骤,从而增增加了反应的选择性,提高了产品质量和收率,适合工业化生产。
具体实施方式
[0033]
以下结合数个较佳实施例对本发明技术方案作进一步非限制性的详细说明。其中所涉及的起始原料2-乙基-2-异硫脲氢溴酸盐(ii)为常见化学试剂,也可参见文献“journal of medicinal chemistry”,49(23),6650-6651;2006对相同化合物的制备方法进行合成。
[0034]
实施例一:
[0035]
室温下,于反应瓶中加入三光气(29.6g,0.1mol)和二氯甲烷(200ml),搅拌下滴加1-甲基-1h-1,2,4-三唑-3-甲基胺(11.2g,0.1mol)和二氯甲烷200ml的溶液。滴加完备后,降温至0~5℃,搅拌下继续滴加三乙胺(15.2g,0.15mol),缓慢升至室温,继续搅拌4小时。减压浓缩,得油状物1-甲基-3-异氰酸酯甲基-1h-1,2,4-三唑(iii)12.6g,收率为91.3%,ei-ms m/z:139[m h]
。
[0036]
实施例二:
[0037]
于反应瓶中加入2-乙基-2-异硫脲氢溴酸盐(ii)(9.25g,50mmol)、1-甲基-3-异氰酸酯甲基-1h-1,2,4-三唑(iii)(7.94g,57.5mmol)、1,8-二氮杂双环[5.4.0]-十一-7-烯(8.75g,57.5mmol)和dmf(100ml),冰浴下搅拌6~8小时。向反应体系中加入n,n
’‑
羰基二咪唑(简称cdi)(9.73g, 60mmol)和1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)(11.42g,75mmol),升至室温搅拌3~4小时,tlc检测反应完成。冰浴降温至0~5℃,滴加稀盐酸至中性,有固体析出,静置析晶后过滤,滤饼用乙酸乙酯重结晶得类白色固体6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(iv)10.2g,收率为76.0%,ei-ms m/z:269[m h]
;1h nmr(dmso d6)δ7.90(s,1h),5.20(s,2h),3.81(s,3h),3.15(q,j=7.4hz,2h),1.30(t,j=7.4hz,3h)。
[0038]
实施例三:
[0039]
于三口瓶中将6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(iv)(5.73g,20mmol)和2,4,5-三氟苄基溴(6.75g,30mmol)溶于乙腈(100ml)中,加入缚酸剂碳酸铯(16.3g,50mmol),升温至回流,反应2~4小时。冷却至室温,用冰水淬灭反应,乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,减压蒸馏回收溶剂,残
余物用乙酸乙酯重结晶得白色固体6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(v)7.85g,收率为94.8%,ei-ms m/z:413[m h]
;1h nmr(dmso d6)δ7.93(s,1h),7.14(m,1h),6.95(m,1h),5.23(s,2h),5.16(s,2h),3.84(s,3h),3.20(q,,j=7.4hz,2h),1.34(t,j=7.4hz,3h)。
[0040]
实施例四:
[0041]
于反应瓶中加入6-乙巯基-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(v)(4.14g,10mmol)、6-氯-2-甲基-2h-吲唑-5-胺(2.72g,15mmol)、醋酸(6.0g,0.1mol)和叔丁醇50ml,回流下搅拌反应6~8小时,tlc检测反应完成。冷却至室温,将反应液倾至饱和碳酸钠水溶液,用乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,减压蒸馏回收溶剂,所得残余物用乙醇重结晶得浅棕色固体(6e)-6-[(6-氯-2-甲基-2h-吲唑-5-基)亚氨基]二氢-3-[(1-甲基-1h-1,2,4-三唑-3-基)甲基]-1-[(2,4,5-三氟苯基)甲基]-1,3,5-三嗪-2,4(1h,3h)-二酮(s-217622,i)4.10g,收率为77.1%,ei-ms m/z:532[m h]
;1h nmr(dmso d6)δ9.31(s,1h),8.40(s,1h),7.73(s,1h),7.53(m,1h),7.67(m,1h),7.44(m,1h),5.26(s,2h),5.04(s,2h),4.15(s,3h),3.90(s,3h)。
[0042]
需要指出的是,上述较佳实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。