1.本发明涉及被膜形成用组合物和被膜以及药液。
背景技术:
2.目前,提出了各种能够对对象物的表面赋予超拨水性的被膜。例如,在专利文献1中,公开了通过使用具有聚合性基团的微粒和在分子内具有2个以上的聚合性基团的化合物,能够形成兼顾超拨水性和耐磨耗性的超拨水性被膜。在专利文献2中,公开了通过适当控制被膜的各性能而能够形成包含氟原子且兼顾超拨水性和耐磨耗性的超拨水性被膜的技术。
3.另外,在专利文献3中,公开了一种包含含有氟代烯烃、含全氟烷基的单体、含有羟基的不饱和单体的氟系共聚物且还包含二氧化硅微粉末的被膜形成用的组合物。据称,采用这样的组合物能够形成拨水拨油性、脱模性、剥离性、保存稳定性优异的被膜。
4.现有技术文献
5.专利文献
6.专利文献1:国际公开第2016/056663号
7.专利文献2:国际公开第2017/179678号
8.专利文献3:日本特开2011-84745号公报
技术实现要素:
9.发明所要解决的课题
10.本发明的目的在于提供一种能够形成具有优异的拨水性、硬度也高、并且耐水性也优异的被膜的被膜形成用组合物和被膜以及药液。
11.用于解决课题的方案
12.即,本发明例如包括以下的项所述的主题。
13.项1
14.一种被膜形成用组合物,其含有无机微粒、聚合性成分和拨水性成分,
15.上述无机微粒的平均粒径为100nm以上5μm以下,
16.上述无机微粒、上述聚合性成分和上述拨水性成分的总质量每100质量份中,上述无机微粒的含量为5~40质量份,
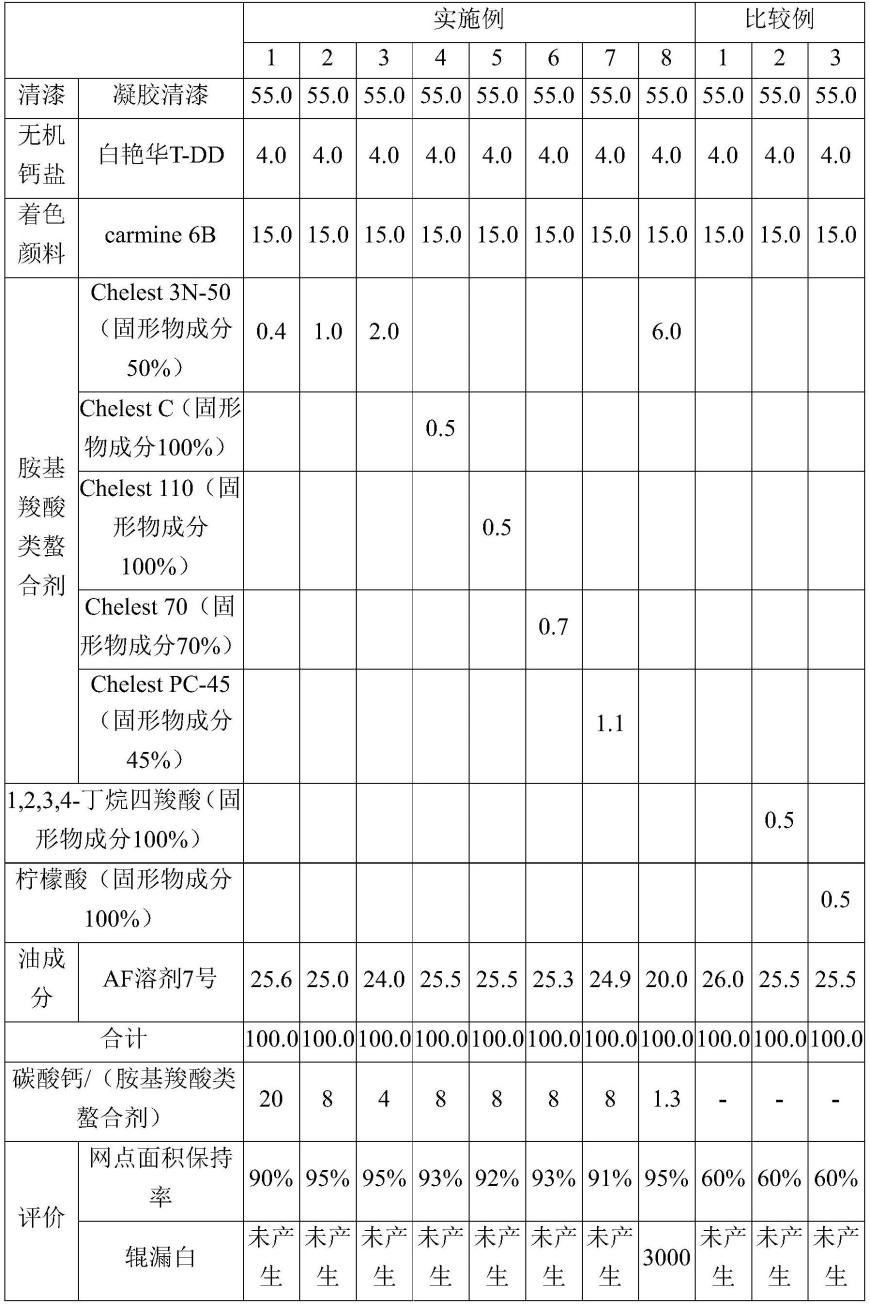
17.上述拨水性成分是至少具有基于下述通式(1)所示的化合物的结构单元的聚合物,
[0018][0019]
(式中,x为氢原子、氟原子、氯原子、溴原子、碘原子、cfx1x2基(其中,x1和x2相同或不同,为氢原子、氟原子或氯原子。)、氰基、碳原子数1~6的直链状或支链状的氟代烷基、取
代或非取代的苄基、取代或非取代的苯基、或者碳原子数1~20的直链状或支链状烷基,y为价键、可以具有氧原子的碳原子数1~10的烃基、-ch2ch2n(r1)so2-基(其中,r1为碳原子数1~4的烷基,式子的右端与ra结合且左端与o结合。)、-ch2ch(oy1)ch2-基(其中,y1为氢原子或乙酰基,式子的右端与ra结合且左端与o结合。)、或-(ch2)nso2-基(n为1~10,式子的右端与ra结合且左端与o结合。),ra为碳原子数20以下的直链状或支链状的烷基、碳原子数6以下的直链状或支链状的氟代烷基、或者分子量400~5000的聚醚基或分子量400~5000的氟代聚醚基。)。
[0020]
项2
[0021]
如项1所述的被膜形成用组合物,其中,在上述无机微粒、上述聚合性成分和上述拨水性成分的总质量每100质量份中,上述拨水性成分的含量为0.1~10质量份。
[0022]
项3
[0023]
如项1或2所述的被膜形成用组合物,其中,上述聚合性成分包含具有交联性官能团的聚合物和固化剂。
[0024]
项4
[0025]
一种包含项1~3中任一项所述的被膜形成用组合物的固化物的被膜。
[0026]
项5
[0027]
一种被膜,其包含无机微粒、粘合剂成分和拨水性成分,
[0028]
上述无机微粒的平均粒径为100nm以上5μm以下,
[0029]
在上述无机微粒、上述粘合剂成分和上述拨水性成分的总质量每100质量份中,上述无机微粒的含量为5~40质量份,
[0030]
上述拨水性成分是至少具有基于下述通式(1)所示的化合物的结构单元的聚合物,
[0031][0032]
(式中,x为氢原子、氟原子、氯原子、溴原子、碘原子、cfx1x2基(其中,x1和x2相同或不同,为氢原子、氟原子或氯原子。)、氰基、碳原子数1~6的直链状或支链状的氟代烷基、取代或非取代的苄基、取代或非取代的苯基、或者碳原子数1~20的直链状或支链状烷基,y为价键、可以具有氧原子的碳原子数1~10的烃基、-ch2ch2n(r1)so2-基(其中,r1为碳原子数1~4的烷基,式子的右端与ra结合且左端与o结合。)、-ch2ch(oy1)ch2-基(其中,y1为氢原子或乙酰基,式子的右端与ra结合且左端与o结合。)、或-(ch2)nso2-基(n为1~10,式子的右端与ra结合且左端与o结合。),ra为碳原子数20以下的直链状或支链状的烷基、碳原子数6以下的直链状或支链状的氟代烷基、或者分子量400~5000的聚醚基或分子量400~5000的氟代聚醚基。)。
[0033]
项6
[0034]
如项5所述的被膜,其中,在上述无机微粒、上述粘合剂成分和上述拨水性成分的总质量每100质量份中,上述拨水性成分的含量为0.1~10质量份。
[0035]
项7
[0036]
一种包含无机微粒的被膜,其中,
[0037]
上述无机微粒的平均粒径为100nm以上5μm以下,
[0038]
水的接触角为150
°
以上,
[0039]
铅笔硬度为hb以上。
[0040]
项8
[0041]
如项7所述的被膜,其还包含不具有氟原子的拨水成分。
[0042]
项9
[0043]
如项7或8所述的被膜,其中,以负荷100g/cm 2
将擦拭纸涂擦至少50次之后的水的接触角为150
°
以上。
[0044]
项10
[0045]
一种包含项1~3中任一项所述的被膜形成用组合物的药液。
[0046]
发明的效果
[0047]
本发明的被膜形成用组合物能够形成具有优异的拨水性、硬度也高、并且耐水性也优异的被膜。
[0048]
本发明的被膜具有优异的拨水性、硬度也高、并且耐水性也优异。
具体实施方式
[0049]
超拨水性被膜例如在纯水中暴露一段时间时,会存在拨水性易于降低的问题,因此,为了长时间维持高的性能,要求提高超拨水性被膜的耐水性。
[0050]
本发明的发明人鉴于上述状况,为了形成具有优异的拨水性、硬度也高、并且耐水性也优异的被膜,反复进行了深入研究。其结果发现,通过组合特定的无机微粒、聚合性成分和拨水性成分,能够实现上述目的。以下,对本发明的实施方式详细地进行说明。
[0051]
此外,在本说明书中,关于“含有”和“包含”的表达,包括“含有”、“包含”、“实质上由
……
构成”和“仅由
……
构成”的概念。
[0052]
在本说明书中,使用“~”表示的数值范围表示作为最小值和最大值分别包含“~”的前后记载的数值的范围。在本说明书中分段记载的数值范围中,某个分段的数值范围的上限值或下限值能够与其它分段的数值范围的上限值或下限值任意组合。在本说明书中所记载的数值范围中,其数值范围的上限值或下限值可以替换为实施例所示的值或能够从实施例毫无疑义地导出的值。
[0053]
1.被膜形成用组合物
[0054]
本发明的被膜形成用组合物至少含有无机微粒、聚合性成分和拨水性成分。
[0055]
(无机微粒)
[0056]
在本发明的被膜形成用组合物中,无机微粒的平均粒径为100nm以上5μm以下。在被膜形成用组合物中,无机微粒的平均粒径的测定方法按照以下所述的步骤。通过加热处理(300℃、3小时)除去被膜形成用组合物的挥发成分,通过扫描型电子显微镜直接观察所得到的微粒,选择200个拍摄画面中的微粒,将对它们的圆当量直径算出的平均值作为被膜形成用组合物中的无机微粒的平均粒径。
[0057]
只要无机微粒的平均粒径为100nm以上5μm以下即可,其种类没有特别限定,例如,能够广泛例示公知的金属氧化物的微粒。具体而言,作为金属氧化物,能够列举二氧化硅、氧化铝、氧化钛、氧化锆等,从容易地提高被膜的硬度和耐水性的方面考虑,优选无机微粒
包含二氧化硅。无机微粒可以仅为二氧化硅。
[0058]
无机微粒的平均粒径低于100nm或超过5μm时,无法发挥所期望的性能。无机微粒的平均粒径更优选为1μm以上3μm以下。
[0059]
无机微粒的比表面积没有特别限定,例如,从容易提高所得到的被膜的硬度的方面考虑,优选为30~700m 2
/g,更优选为100~300m 2
/g。
[0060]
在本发明的被膜形成用组合物中,上述微粒的比表面积是指通过bet法测得的值(所谓的bet比表面积)。
[0061]
无机微粒的表面可以为亲水性的,也可以为疏水性的。本发明的被膜形成用组合物所含的无机微粒也可以为亲水性的无机微粒和疏水性的无机微粒的混合物。
[0062]
无机微粒的形状也没有特别限定,例如,能够列举球状、椭圆球状等,另外,也可以为异形状等的不定形颗粒。
[0063]
(拨水性成分)
[0064]
在本发明的被膜形成用组合物中,拨水性成分是在被膜形成时能够对被膜赋予拨水性的成分。拨水性成分包含具有基于下述通式(1)所示的化合物的结构单元的聚合物。
[0065][0066]
上述式(1)中、x表示氢原子、氟原子、氯原子、溴原子、碘原子、cfx1x2基(其中,x1和x2相同或不同,为氢原子、氟原子或氯原子。)、氰基、碳原子数1~6的直链状或支链状的氟代烷基、取代或非取代的苄基、取代或非取代的苯基、或者碳原子数1~20的直链状或支链状烷基。x为碳原子数3以上的烷基或氟代烷基时,它们可以为环状或非环状的任意一种。另外,x为烷基时,其碳原子数优选为1~10、更优选为1~6、进一步优选为1~2。另外,为氟代烷基时,其碳原子数优选为1~6、更优选为1~4、进一步优选为1~2。
[0067]
上述式(1)中,y表示价键、可以具有氧原子的碳原子数1~10的烃基、-ch2ch2n(r1)so2-基(其中,r1为碳原子数1~4的烷基,式子的右端与ra结合且左端与o结合。)、-ch2ch(oy1)ch2-基(其中,y1为氢原子或乙酰基,式子的右端与ra结合且左端与o结合。)、或-(ch2)nso2-基(n为1~10,式子的右端与ra结合且左端与o结合。)。“价键”是指在上述式(1)中y的两端的ra与o直接结合,即y不包含元素。y为碳原子数1~10的烃基时,具体而言,为碳原子数1~10的亚烷基,优选为碳原子数1~6的亚烷基,进一步优选为碳原子数1~2的亚烷基。
[0068]
上述式(1)中,ra表示碳原子数20以下的直链状或支链状的烷基、碳原子数6以下的直链状或支链状的氟代烷基、或者分子量400~5000的聚醚基或分子量400~5000的氟代聚醚基。氟代烷基例如优选为全氟烷基,氟代聚醚基例如优选为全氟聚醚基。
[0069]
作为式(1)所示的化合物的具体例,例如,能够广泛列举上述专利文献1中公开的丙烯酸酯。
[0070]
其中,从所形成的被膜的拨水性和拨油性优异、耐水性也容易提高的观点考虑,式(1)所示的化合物可以列举:x为氢原子、氯原子、氟原子或甲基、y为价键或碳原子数1~10的亚烷基(优选为碳原子数1~6的亚烷基、进一步优选为碳原子数1~2的亚烷基)、ra为碳原子数1~20的直链状或支链状的烷基、碳原子数1~6的直链状或支链状的氟代烷基、或者
分子量400~5000的聚醚基或分子量400~5000的氟代聚醚基的组合。更优选的式(1)所示的化合物中,x为氢原子或甲基、y为碳原子数1~2的亚烷基、ra为碳原子数1~6的直链状或支链状的氟代烷基、或者x为氢原子或甲基、y为价键、ra为碳原子数1~20的直链状或支链状的烷基。
[0071]
作为式(1)所示的化合物的具体化合物,可以列举氟代烷基的碳原子数为1~6的(甲基)丙烯酸氟代烷基酯和烷基的碳原子数为1~20(优选为1~18)的(甲基)丙烯酸烷基酯。
[0072]
拨水性成分在式(1)所示的化合物以外,还可以包含基于具有环氧部位的(甲基)丙烯酸酯的结构单元、或基于具有酰胺键的(甲基)丙烯酸酯(例如,上述式(1)的y替换为具有酰胺键的官能团的化合物)的结构单元。例如,作为具有环氧部位的(甲基)丙烯酸酯,能够列举(甲基)丙烯酸缩水甘油酯,作为具有酰胺键的(甲基)丙烯酸酯,能够列举硬脂酸酰胺(甲基)丙烯酸乙酯。
[0073]
作为拨水性成分的具体例,能够列举主骨架为(甲基)丙烯酸酯且在其侧链部位具有氟代烷基或烷基的聚合物。拨水性成分例如能够以公知的方法进行制造。或者,拨水性成分也能够从市售品获得,例如,能够列举大金工业制的商品名“unidyne tg series”。
[0074]
(聚合性成分)
[0075]
在本发明的被膜形成用组合物中,聚合性成分是形成被膜时成为后述粘合剂成分的成分,能够称为粘合剂成分前体。即,聚合性成分是能够固化(例如,聚合反应或固化反应)的成分,是固化后形成被膜的粘合剂成分的成分。
[0076]
聚合性成分只要能够形成粘合剂成分即可,其种类没有特别限定,能够广泛使用公知的材料。例如,作为被膜形成用组合物中所含的聚合性成分,能够列举通过赋予热而进行固化反应从而能够形成固化物的成分、或者能够通过uv等的光照射而形成固化物的成分。将通过赋予热而进行固化反应从而能够形成固化物的成分记为“热固性树脂成分”,将能够通过光照射而形成固化物的成分记为“光固性树脂成分”。
[0077]
热固性树脂成分的种类没有特别限定,例如,能够为公知的聚合物和根据需要含有的固化剂的混合物。这样的聚合物例如优选具有交联性官能团,此时,例如,容易通过热固化进行交联反应。根据该观点,聚合性成分(热固性树脂成分)优选含有具有交联性官能团的聚合物和固化剂。
[0078]
热固性树脂成分中所含的聚合物可以具有氟原子(氟代烷基),也可以不具有氟原子(氟代烷基)。但是,从容易提高被膜的拨水性的观点考虑,热固性树脂成分中所含的聚合物优选具有氟代烷基。
[0079]
热固性树脂成分中所含的聚合物例如优选为通过后述的固化剂和热而反应从而进行固化反应的化合物。作为例子,可以列举上述的具有交联性官能团的聚合物。交联性官能团例如优选为羟基、羧基、氨基、硫醇基、异氰酸酯基等。此时,这样的化合物优选还具有氟代烷基。以下,将具有交联性官能团的聚合物简记为“交联性聚合物”。
[0080]
从这样的观点考虑,作为交联性聚合物,优选应用具有反应性官能团的含氟聚合物。作为含氟聚合物,例如,能够列举包含如下结构单元的聚合物:
[0081]
(a)四氟乙烯结构单元;
[0082]
(b)不含羟基和羧基的非芳香族系的乙烯酯单体结构单元;
[0083]
(c)不含芳香族基团和羧基的含羟基的乙烯基单体结构单元;
[0084]
(e)不含羟基和芳香族基团的含羧基的单体结构单元;和
[0085]
(f)其它单体结构单元(其中,不含(d)不含羟基和羧基的含芳香族基团的单体结构单元)。
[0086]
以下,将该聚合物记为“聚合物f”。热固性树脂为聚合物f时,特别能够提高被膜的耐水性。
[0087]
关于上述(a)四氟乙烯结构单元的含有比例,在聚合物f的总量中,下限为20摩尔%、优选为30摩尔%、更优选为40摩尔%、特别优选为42摩尔%,上限为49摩尔%、优选为47摩尔%。
[0088]
作为形成上述非芳香族系的乙烯酯单体结构单元(b)的单体,例如,可以列举乙酸乙烯酯、丙酸乙烯酯、丁酸乙烯酯、异丁酸乙烯酯、特戊酸乙烯酯、己酸乙烯酯、叔碳酸乙烯酯(vinyl versatate)、月桂酸乙烯酯、硬脂酸乙烯酯、环己酸乙烯酯等的1种或2种以上。这些单体是不含羟基和羧基的非芳香族系单体。从耐候性等优异的方面考虑,特别优选的形成非芳香族系的乙烯酯单体结构单元(b)的单体为选自叔碳酸乙烯酯、月桂酸乙烯酯、硬脂酸乙烯酯、环己酸乙烯酯和乙酸乙烯酯中的1种。这些之中,从耐药品性方面考虑,优选为非芳香族系羧酸乙烯酯、特别是羧酸的碳原子数为6以上的羧酸乙烯酯、进一步优选为羧酸的碳原子数为9以上的羧酸乙烯酯。羧酸乙烯酯中的羧酸的碳原子数的上限优选为20以下、进一步优选为15以下。作为具体例,最优选为叔碳酸乙烯酯。
[0089]
关于上述非芳香族系的乙烯酯单体结构单元(b)的含有比例,在聚合物f的总量中,下限为25摩尔%、优选为30摩尔%,上限为69.9摩尔%、优选为60摩尔%、更优选为43摩尔%、特别优选为40摩尔%。
[0090]
形成上述含羟基的乙烯基单体结构单元(c)的单体为不含羧基的非芳香族系的单体,例如可以列举式(2)所示的羟基烷基乙烯基醚或羟基烷基烯丙基醚。
[0091]
ch2=chr
10
ꢀꢀꢀ
(2)
[0092]
其中,式(2)中,r
10
表示-or
20
或-ch2or
20
(其中,r
20
为具有羟基的烷基。)。作为r
20
,例如可以列举在碳原子数1~8的直链状或支链状的烷基结合有1~3个、优选结合有1个羟基的基团。作为式(2)的例子,可以列举2-羟基乙基乙烯基醚、3-羟基丙基乙烯基醚、2-羟基丙基乙烯基醚、2-羟基-2-甲基丙基乙烯基醚、4-羟基丁基乙烯基醚、4-羟基-2-甲基丁基乙烯基醚、5-羟基戊基乙烯基醚、6-羟基己基乙烯基醚、2-羟基乙基烯丙基醚、4-羟基丁基烯丙基醚、甘油单烯丙基醚等的1种或2种以上。其中,形成上述含羟基的乙烯基单体结构单元(c)的单体优选为4-羟基丁基乙烯基醚、2-羟基乙基乙烯基醚。
[0093]
通过该含羟基的乙烯基单体结构单元(c)的存在,能够改善被膜的加工性、耐冲击性、耐污染性。
[0094]
关于含羟基的乙烯基单体结构单元(c)的含有比例,在聚合物f的总量中,下限为8摩尔%、优选为10摩尔%、进一步优选为15摩尔%,上限为30摩尔%、优选为20摩尔%。
[0095]
聚合物f能够基本上由(a)、(b)和(c)(其中,各单元之内可以2种以上共聚)构成,能够含有直至10摩尔%的其它能够共聚的单体单元(f)。其它能够共聚的单体结构单元(f)为上述(a)、(b)和(c)以外的含芳香族基团的单体结构单元(d)和含羧基的单体结构单元(e)以外的单体结构单元。
[0096]
作为形成其它能够共聚的单体结构单元(f)的单体,例如,可以列举甲基乙烯基醚、乙基乙烯基醚等的烷基乙烯基醚;乙烯、丙烯、正丁烯、异丁烯等非氟系的烯烃等。含有其它能够共聚的单体结构单元(f)时,其含有比例在聚合物f中为10摩尔%以下、优选低于5摩尔%、进一步优选为4摩尔%以下。
[0097]
聚合物f还能够含有(d)不含羟基和羧基的含芳香族基团的单体结构单元。作为(d)不含羟基和羧基的含芳香族基团的单体结构单元,例如,可以列举苯甲酸乙烯酯、对叔丁基苯甲酸乙烯酯等的苯甲酸乙烯基单体等的1种或2种以上,特别优选对叔丁基苯甲酸乙烯酯、进一步优选苯甲酸乙烯酯。
[0098]
关于含芳香族基团的单体结构单元(d)的含有比例,在聚合物f中,下限为2摩尔%、优选为4摩尔%,上限为15摩尔%、优选为10摩尔%、更优选为8摩尔%。
[0099]
聚合物f还能够含有(e)不含羟基和芳香族基团的含羧基的单体结构单元。作为含羧基的单体,例如,可以列举丙烯酸、甲基丙烯酸、乙烯基乙酸、巴豆酸、肉桂酸、3-烯丙氧基丙酸、衣康酸、衣康酸单酯、马来酸、马来酸单酯、马来酸酐、富马酸、富马酸单酯、苯二甲酸乙烯酯、均四苯酸乙烯酯等的1种或2种以上。其中,优选均聚性低的巴豆酸、衣康酸、马来酸、马来酸单酯、富马酸、富马酸单酯、3-烯丙氧基丙酸。
[0100]
在聚合物f中,含羧基的单体结构单元(e)的含有比例的下限为0.1摩尔%、优选为0.4摩尔%,上限为2.0摩尔%、优选为1.5摩尔%。
[0101]
聚合物f通过使用四氢呋喃作为洗脱液的凝胶色谱层析(gpc)测定的数均分子量例如为1000~1000000、优选为3000~50000。通过差示扫描量热计(dsc)求得的聚合物f的玻璃化转变温度(2nd run)例如为10~60℃、优选为20~40℃。聚合物f的制造方法也没有特别限定,能够广泛采用公知的制造方法。另外,聚合物f也能够作为市售品等购入。
[0102]
作为聚合物f的具体例,可以列举大金工业株式会社制的zeffle(注册商标)gk系列等。
[0103]
在本发明的被膜形成用组合物中,热固性树脂成分能够如上所述含有固化剂。固化剂没有特别限定,例如,能够广泛应用作为热固性树脂用的固化剂使用的化合物。特别是,优选固化剂是在上述的分子内具有2个以上的聚合性基团的化合物。
[0104]
作为固化剂的具体例,可以列举异氰酸酯系固化剂。作为异氰酸酯系固化剂,例如,可以列举具有异氰酸酯基的化合物(以下,简单记为异氰酸酯化合物)。异氰酸酯化合物例如可以列举下述通式(20)所示的化合物。
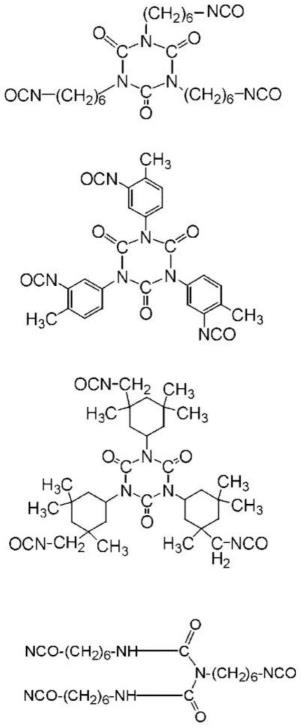
[0105][0106]
式(20)中,z6是在至少一个末端具有异氰酸酯基、可以至少一个碳原子被杂原子取代、可以至少一个氢原子被卤原子取代、可以具有碳-碳不饱和键的、直链状或支链状的1价的烃基或羰基。r3是可以至少一个碳原子被杂原子取代、可以至少一个氢原子被卤原子取代、可以至少一个碳原子被杂原子取代、可以具有碳-碳不饱和键的、支链状或环状的2价以上的烃基或羰基。式(20)中的o为2以上的整数。
[0107]
r3优选为碳原子数1~20,更优选为碳原子数2~15,进一步优选为碳原子数3~10。
[0108]
z6优选为碳原子数1~20,更优选为碳原子数2~15,进一步优选为碳原子数3~
10。
[0109]
异氰酸酯化合物可以使用1种,或者也可以组合使用多种。
[0110]
作为异氰酸酯化合物,例如,能够列举多异氰酸酯。在本说明书中,多异氰酸酯是指在分子内具有2个以上异氰酸酯基的化合物。异氰酸酯化合物可以是通过将二异氰酸酯三聚体化得到的多异氰酸酯。这样的通过将二异氰酸酯三聚体化得到的多异氰酸酯可以是三异氰酸酯。作为二异氰酸酯的三聚体的多异氰酸酯可以作为将它们聚合得到的聚合物存在。
[0111]
作为二异氰酸酯,没有特别限定,可以列举三亚甲基二异氰酸酯、六亚甲基二异氰酸酯、异佛尔酮二异氰酸酯、二甲苯二异氰酸酯、氢化二甲苯二异氰酸酯、环己烷二异氰酸酯、二环己基甲烷二异氰酸酯、降冰片烷二异氰酸酯等的异氰酸酯基与脂肪族基团结合的二异氰酸酯;甲苯二异氰酸酯、二苯基甲烷二异氰酸酯、聚亚甲基聚苯基多异氰酸酯、联甲苯胺二异氰酸酯、萘二异氰酸酯等的异氰酸酯基与芳香族基团结合的二异氰酸酯。
[0112]
作为具体的多异氰酸酯,没有特别限定,可以列举具有下述的结构的化合物。
[0113][0114]
这些多异氰酸酯可以作为聚合物存在,例如,为六亚甲基二异氰酸酯的异氰脲酸酯型多异氰酸酯时,可以存在具有下述结构的聚合物。
[0115][0116]
在优选的实施方式中,异氰酸酯化合物为异氰脲酸酯型多异氰酸酯。
[0117]
上述异氰脲酸酯型多异氰酸酯可以为它们聚合得到的聚合物。异氰脲酸酯型多异氰酸酯可以为仅具有1个异氰脲酸酯环的单环式化合物,或者也可以是该单环式化合物聚合得到的多环式化合物。
[0118]
在使用两种以上的异氰酸酯化合物的一个方式中,能够使用包含仅具有1个异氰脲酸酯环的单环式化合物的混合物。
[0119]
在使用两种以上的异氰酸酯化合物的另外的方式中,能够使用包含作为异氰脲酸酯型多异氰酸酯的异氰酸酯化合物的混合物。异氰脲酸酯型多异氰酸酯例如可以为三异氰酸酯,具体而言,可以为通过将二异氰酸酯三聚体化得到的三异氰酸酯。
[0120]
作为异氰酸酯化合物的具体例,例如,可以列举2,4-甲苯二异氰酸酯、二苯基甲烷-4,4’-二异氰酸酯、二甲苯二异氰酸酯、异佛尔酮二异氰酸酯、赖氨酸甲酯二异氰酸酯、甲基环己基二异氰酸酯、三甲基六亚甲基二异氰酸酯、六亚甲基二异氰酸酯、正戊烷-1,4-二异氰酸酯、它们的三聚体、它们的加成物、缩二脲体、异氰脲酸酯体、这些聚合物中具有2个以上的异氰酸酯基的化合物、进一步进行了封端的异氰酸酯类等。作为更详细的固化剂的具体例,能够使用作为sumidur(注册商标)n3300(住化covestro polyurethane株式会社制)、desmodur(注册商标)n3600(住化covestro polyurethane株式会社制)、desmodur t、l、il、hl系列(住化covestro polyurethane株式会社制)、desmodur(注册商标)2460m(住化covestro polyurethane株式会社制)、sumidur(注册商标)44系列(住化covestro polyurethane株式会社制)、sbu异氰酸酯系列(住化covestro polyurethane株式会社制)、desmodur(注册商标)e、m系列(住化covestro polyurethane株式会社制)、sumidur ht(住化covestro polyurethane株式会社制)、desmodur n系列(住化covestro polyurethane株式会社制)、desmodur z4470系列(住化covestro polyurethane株式会社制)、duranate tpa-100(旭化成株式会社制)、duranate tka-100(旭化成株式会社制)、duranate 24a-100(旭化成株式会社制)、duranate 22a-75p(旭化成株式会社制)和duranate p301-75e(旭化成株式会社制)市售的固化剂等。
[0121]
热固性树脂成分含有固化剂时,其含有比例相对于热固性树脂成分的总质量能够为10~100质量%,优选为15~35质量%。
[0122]
另一方面,聚合性成分为光固性树脂成分时,只要是能够利用uv等的光照射进行聚合反应的单体即可,其种类没有特别限定,例如,能够广泛采用公知的光聚合性单体。聚合性成分为光固性树脂成分时,例如,能够使用公知的光聚合引发剂。
[0123]
(被膜形成用组合物)
[0124]
在本发明的被膜形成用组合物中,只要不损及本发明的效果,无机微粒、聚合性成
分和拨水性成分的含有比例没有特别限定。
[0125]
在本发明的被膜形成用组合物中,相对于上述无机微粒、上述聚合性成分和上述拨水性成分的总质量100质量份(固体成分换算),无机微粒的含量为5~40质量份。无机微粒的含量超出该范围时,被膜的硬度降低。相对于上述无机微粒、上述聚合性成分和上述拨水性成分的总质量100质量份(固体成分换算),无机微粒的含量更优选为20~40质量份。
[0126]
在本发明的被膜形成用组合物中,相对于上述无机微粒、上述聚合性成分和上述拨水性成分的总质量100质量份(固体成分换算),聚合性成分的含量优选为50~90质量份。此时,被膜的硬度不易降低,拨水性和耐水性容易提高。相对于上述无机微粒、上述聚合性成分和上述拨水性成分的总质量100质量份(固体成分换算),聚合性成分的含量更优选为60~80质量份。
[0127]
例如,在本发明的被膜形成用组合物中,相对于上述无机微粒、聚合性成分和上述拨水性成分的总质量100质量份(固体成分换算),拨水性成分的含量优选为0.1~10质量份,更优选为1~5质量份。
[0128]
本发明的被膜形成用组合物中,优选上述聚合性成分和上述拨水性成分的至少一方具有氟原子(即,氟代烷基),更优选上述聚合性成分具有氟原子。由此,由被膜形成用组合物形成的被膜具有优异的拨水性、硬度也高、并且耐水性也优异,还能够提高耐磨耗性。当然,也能够上述聚合性成分和上述拨水性成分双方均具有氟原子(氟代烷基)。作为上述聚合性成分具有氟代烷基的情况的一例,可以列举上述热固性树脂成分所含的交联性聚合物为聚合物f的情况。作为上述拨水性成分具有氟代烷基的情况的一例,可以列举在式(1)所示的化合物中氟代烷基的碳原子数为1~6的氟代烷基(甲基)丙烯酸酯的聚合物。
[0129]
本发明的被膜形成用组合物所含的拨水性成分不具有氟原子(特别是氟代烷基)时,容易提高被膜的耐磨耗性。其中,在拨水性成分中所含的上述式(1)所示的化合物中,ra为碳原子数20以下的直链或支链状的烷基时,更容易提高被膜的耐磨耗性。作为这样的化合物,、例如,能够列举公知的润滑油所含的饱和脂肪酸等,作为其例子,可以列举椰油、棕榈油、菜籽油、蓖麻油等所含的辛酸、癸酸、月桂酸、肉豆蔻酸、棕榈酸、硬脂酸或具有与这些饱和脂肪酸同样的碳原子数的直链烷基的结构体。
[0130]
本发明的被膜形成用组合物只要包含无机微粒、聚合性成分和拨水性成分即可,也可以包含其它成分。例如,被膜形成用组合物中也能够根据需要包含溶剂。溶剂的种类没有限定,例如,能够广泛使用用于形成被膜的溶剂,例如,能够列举氢氟醚等的氟系溶剂、酯化合物、醇化合物等。
[0131]
被膜形成用组合物中所含的溶剂的含量没有特别限定,例如,在无机微粒、聚合性成分和拨水性成分的总质量每100质量份中,能够设为200~2000质量份。
[0132]
被膜形成用组合物也能够包含溶剂以外的其它添加剂。被膜形成用组合物包含其它添加剂时,其含有比例期望相对于无机微粒、聚合性成分和拨水性成分的总质量为5质量%以下。
[0133]
被膜形成用组合物此外例如还能够包含有机微粒等,为了表现出被膜的硬度,被膜形成用组合物优选不含有机微粒。
[0134]
本发明的被膜形成用组合物的制备方法没有特别限定。例如,能够通过以规定的配合量混合无机微粒、聚合性成分(例如,包含上述聚合物f和固化剂的热固性树脂成分)和
拨水性成分来制备。混合方法也没有特别限定,例如,能够广泛使用公知的混合机等。
[0135]
使用本发明的被膜形成用组合物形成被膜的方法没有特别限定。例如,通过将被膜形成用组合物涂布于用于形成被膜的基材,形成涂膜,将该涂膜固化,能够形成被膜。这样的被膜为本发明的被膜形成用组合物的固化物。
[0136]
被膜形成用组合物的涂布方法没有特别限制,能够广泛采用公知的涂膜形成方法。例如,能够通过刷涂、喷雾、旋涂、分配器等的方法涂布。用于形成被膜形成用组合物的涂膜的基材的种类没有特别限定,例如,能够列举丙烯酸树脂、聚碳酸酯树脂等的公知的树脂基材、以及金属、无机基材等的各种基材。
[0137]
被膜形成用组合物的固化方法也没有特别限定,例如,能够采用热固化、光固化等各种方法,也能够组合热固化和光固化。采用热固化时,优选将被膜形成用组合物的涂膜加热到60~150℃,此时,容易形成被膜形成用组合物的被膜,硬度也高,耐水性也优异。加热时间没有特别限制,能够根据加热温度适当设定。
[0138]
能够使用本发明的被膜形成用组合物制备药液。这样的药液包含本发明的被膜形成用组合物,因此,适用于形成被膜。药液能够仅由被膜形成用组合物构成,或者,只要是不损及成为本发明的被膜形成用组合物的目的的各种性能的程度,能够包含其它添加剂(例如,作为被膜形成溶液、药剂使用的公知的添加剂)。
[0139]
2.被膜
[0140]
本发明的被膜例如能够使用上述的本发明的被膜形成用组合物形成,或者,本发明的被膜能够使用上述药液形成。此时,本发明的被膜为本发明的被膜形成用组合物或药液的固化物。
[0141]
因此,本发明的被膜包含无机微粒、粘合剂成分和拨水性成分。
[0142]
在本发明的被膜中,无机微粒与本发明的被膜形成用组合物中所含的无机微粒相同,因此,其平均粒径为100nm以上5μm以下。
[0143]
其中,被膜中的无机微粒的平均粒径根据以下的步骤测定。首先通过对被膜进行加热处理,能够将被膜中的有机成分烧掉,由此,将被膜中的微粒分离。加热处理在大气氛围下在300℃进行3小时。通过扫描型电子显微镜直接观察所得到的微粒,选择200个拍摄图像中的微粒,算出它们的圆当量直径,将平均值作为被膜中的微粒的平均粒径。
[0144]
在本发明的被膜中,粘合剂成分是使上述的聚合性成分固化形成的成分。例如,聚合性成分为上述的热固性树脂成分时,上述聚合物f是由上述固化剂固化得到(交联得到)的聚合物。粘合剂成分中可以在不损及本发明的被膜的性能的范围混合有未固化的聚合性成分。
[0145]
在本发明的被膜中,拨水性成分与上述的本发明的被膜形成用组合物中所含的拨水性成分相同。因此,被膜中的拨水性成分包含具有来自上述式(1)所示的化合物的结构单元的聚合物,另外,拨水性成分可以包含式(1)所示的化合物以外的、基于具有环氧部位的(甲基)丙烯酸酯的结构单元或基于具有酰胺键的(甲基)丙烯酸酯(例如,上述式(1)的y替换为具有酰胺键的官能团的化合物)的结构单元。例如,作为具有环氧部位的(甲基)丙烯酸酯,能够列举(甲基)丙烯酸缩水甘油酯,作为具有酰胺键的(甲基)丙烯酸酯,能够列举硬脂酸酰胺乙基(甲基)丙烯酸酯。
[0146]
在本发明的被膜中,无机微粒的含量在上述无机微粒、上述粘合剂成分和上述拨
水性成分的总质量每100质量份中为5~40质量份。无机微粒的含量超出该范围时,被膜的硬度降低。无机微粒的含量在上述无机微粒、上述粘合剂成分和上述拨水性成分的总质量每100质量份中更优选为20~40质量份。
[0147]
在本发明的被膜中,粘合剂成分的含量在上述无机微粒、上述粘合剂成分和上述拨水性成分的总质量每100质量份中优选为50~90质量份。此时,被膜的硬度不易降低,拨水性和耐水性容易提高。粘合剂成分的含量在上述无机微粒、上述粘合剂成分和上述拨水性成分的总质量每100质量份中优选为60~80质量份。
[0148]
例如,在本发明的被膜中,拨水性成分的含量在上述无机微粒、上述粘合剂成分和上述拨水性成分的总质量每100质量份中优选为0.1~10质量份,更优选为1~5质量份。
[0149]
本发明的被膜中,优选上述粘合剂成分和上述拨水性成分的至少一方具有氟原子(即,氟代烷基),更优选上述粘合剂成分具有氟原子。由此,由被膜形成用组合物形成的被膜具有优异的拨水性,硬度也高,并且,耐水性也优异,另外,耐磨耗性提高。另外,被膜中所含的拨水性成分不具有氟代烷基时,该被膜的耐磨耗性容易显著提高。当然,也能够使上述粘合剂成分和上述拨水性成分双方均具有氟原子(氟代烷基)。作为上述粘合剂成分具有氟代烷基的情况下的一例,可以列举聚合物f及其交联体。上述拨水性成分具有氟代烷基时,可以列举在式(1)所示的化合物中氟代烷基的碳原子数为1~6的氟代烷基(甲基)丙烯酸酯的聚合物。
[0150]
本发明的被膜例如水接触角为150
°
以上。由此,本发明的被膜能够发挥优异的拨水性。
[0151]
本发明的被膜例如铅笔硬度为hb以上。
[0152]
本发明的被膜例如具有擦拭纸以负荷100g/cm 2
至少涂擦50次之后的水的接触角为150
°
以上的表面。
[0153]
本发明的被膜例如平均膜厚为10nm~100μm、最低局部膜厚为5nm~50μm、最大局部膜厚为15nm~150μm,优选平均膜厚为15nm~95μm、最低局部膜厚为10nm~45μm、最大局部膜厚为20nm~145μm,更优选平均膜厚为20nm~90μm、最低局部膜厚为15nm~40μm、最大局部膜厚为25nm~140μm。平均膜厚例如能够通过由涂布的涂膜量、固体成分浓度、组合物的密度算出的干燥膜厚来计算。最低局部膜厚、最大局部膜厚能够通过使用能够测定三维形状的装置来计算。作为其装置的一例,可以列举keyence制的形状解析激光显微镜vk-x1000。
[0154]
由本发明的被膜形成用组合物形成的被膜具有优异的拨水性,硬度也高,并且耐水性也优异。特别是,这样的被膜的耐水性优异,因此,即使长期暴露于水或盐水中,与现有的被膜相比,也不易发生拨水性的降低。其结果,由本发明的被膜形成用组合物形成的被膜容易长期维持性能。另外,由本发明的被膜形成用组合物形成的被膜的耐磨耗性也容易变得良好。
[0155]
因此,本发明的被膜用于对被处理面赋予拨水性和/或拨油性,能够适用于要求超拨液性的各种物品等。
[0156]
本发明的被膜的用途没有特别限定,例如,能够对拨水拨油剂、结霜延迟用途、防冰效果剂、防雪效果剂、指纹附着防止剂、指纹不识别化剂、低摩擦剂、润滑剂、蛋白质附着控制剂、细胞附着控制剂、微生物附着控制剂、水垢附着抑制剂、防霉剂、防菌剂等适用本发
明的被膜。
[0157]
3.被膜的其它方式
[0158]
作为其它方式,本发明的被膜还能够包含如下的被膜:包含无机微粒,上述无机微粒的平均粒径为100nm以上5μm以下,水的接触角为150
°
以上,铅笔硬度为hb以上(以下,记为“其它方式的被膜”)。形成这样的其它方式的被膜的方法没有特别限制。例如,在使用上述的本发明的被膜形成用组合物或药液形成的情况下,也能够形成上述其它方式的被膜。
[0159]
其它方式的被膜中,无机微粒也与本发明的被膜形成用组合物中所包含的无机微粒相同。
[0160]
在其它方式的被膜中,只要无机微粒的平均粒径为100nm以上5μm以下即可,其种类没有特别限定,例如,能够广泛例示公知的金属氧化物的微粒。具体而言,作为金属氧化物,能够列举二氧化硅、氧化铝、氧化钛、氧化锆等,从容易提高被膜的硬度的方面考虑,优选无机微粒为二氧化硅。
[0161]
无机微粒的平均粒径低于100nm或超过5μm时,无法发挥所期望的性能。更优选无机微粒的平均粒径为1μm以上3μm以下。
[0162]
在其它方式的被膜中,无机微粒的含量在被膜的总质量每100质量份中为5~40质量份。无机微粒的含量超出该范围时,被膜的硬度降低。更优选无机微粒的含量在上述无机微粒、被膜的总质量每100质量份中为20~40质量份。
[0163]
其它方式的被膜包含上述粘合剂成分时,优选其含量在被膜的总质量每100质量份中为50~90质量份。此时,被膜的硬度不易降低,拨水性和耐水性容易提高。更优选聚合性成分的含量在被膜的总质量每100质量份中为60~80质量份。
[0164]
其它方式的被膜包含上述拨水性成分时,优选其含量在被膜的总质量每100质量份中为0.1~10质量份,更优选为1~5质量份。
[0165]
其它方式的被膜通过使得水接触角为150
°
以上,可以发挥优异的拨水性。水接触角例如能够通过适当选择无机微粒、粘合剂成分、拨水性成分的种类和含量来调节。
[0166]
其它方式的被膜通过使得铅笔硬度为hb以上,而具有优异的硬度。铅笔硬度例如能够通过适当选择无机微粒、粘合剂成分、拨水性成分的种类和含量来调节。
[0167]
其它方式的被膜例如也能够具有擦拭纸以负荷100g/cm 2
至少涂擦50次(优选为50次)后的水的接触角为150
°
以上的表面。由此,其它的被膜具有优异的耐磨耗性。
[0168]
本发明的被膜例如平均膜厚为10nm~100μm、最低局部膜厚为5nm~50μm、最大局部膜厚为15nm~150μm,优选平均膜厚为15nm~95μm、最低局部膜厚为10nm~45μm、最大局部膜厚为20nm~145μm,更优选平均膜厚为20nm~90μm、最低局部膜厚为15nm~40μm、最大局部膜厚为25nm~140μm。平均膜厚例如能够通过由涂布的涂膜量、固体成分浓度、组合物的密度算出的干燥膜厚来计算。最低局部膜厚、最大局部膜厚能够通过使用能够测定三维形状的装置来计算。作为其装置的一例,可以列举keyence制的形状解析激光显微镜vk-x1000。
[0169]
其它方式的被膜例如优选将被膜在室温气氛下静置的10~30℃的水中浸渍72小时后的水接触角为150
°
以上。此时,其它方式的被膜的耐水性优异。这样的水接触角例如能够通过适当选择无机微粒、粘合剂成分、拨水性成分的种类和含量来调节。
[0170]
本发明的其他的被膜的用途没有特别限定,例如,能够对拨水拨油剂、结霜延迟用
途、防冰效果剂、防雪效果剂、指纹附着防止剂、指纹不识别化剂、低摩擦剂、润滑剂、蛋白质附着控制剂、细胞附着控制剂、微生物附着控制剂、水垢附着抑制剂、防霉剂、防菌剂等适用本发明的其它的被膜。
[0171]
(本发明的被膜的各种物性的测定方法)
[0172]
对本发明的被膜(也包括上述的其它方式的被膜)的各种物性的测定方法进行说明。
[0173]
<水的接触角(静态接触角)>
[0174]
水的接触角即水的静态接触角。水的接触角使用接触角计(协和界面科学公司“drop master 701”)进行测定,具体而言,使用水(2μl的液滴),对1个样品进行5点测定。静态接触角为150
°
以上时,该液体有时变得无法自立而在基材表面存在。这样的情况下,将注射器的针头作为支撑体测定静态接触角,将此时得到的值作为静态接触角。
[0175]
<铅笔硬度>
[0176]
铅笔硬度的测定方法根据jis k 5600-5进行。
[0177]
<72h浸水试验后的接触角>
[0178]
72h浸水试验后的接触角是将被膜在室温气氛下静置的10~30℃的水中浸渍72小时后,通过与上述的水的接触角的测定方法同样的测定方法来实施。此时的水接触角也通过接触角计(协和界面科学公司“drop master 701”)测定。
[0179]
<滚落角>
[0180]
滚落角的测定中,作为测定仪器,使用了drop master 701(协和界面科学株式会社制)。在注射针的前端形成20μl的水滴后,使载置于水平的试样载台上的涂敷基板表面与注射针前端的水滴的距离通过试样载台侧的移动而缓慢接近,在两者接触时使试样载台和注射针暂时静止,接着,使试样载台侧移动,缓慢分离,由此,使水滴在涂敷基板表面着滴。着滴后,在大概5秒以内使试样载台以2
°
/秒的倾斜速度倾斜,对倾斜角每1
°
以变焦倍率“w1”拍摄基板表面的水滴图像的静止画面。将水滴的后退侧的接触线开始移动时(移动0.1~1mm时)的试样载台的倾斜角度作为滚落角。
[0181]
<滚落速度>
[0182]
滚落速度的测定中,作为测定仪器,使用drop master 701(协和界面科学株式会社制)。在注射针的前端形成20μl的水滴后,使载置于预先倾斜30
°
的试样载台上的涂敷基板表面与注射针前端的水滴的距离通过试样载台侧的可动而缓慢接近,在两者接触时使试样载台和注射针暂时静止。在该时刻,水滴通过注射针在倾斜的涂敷基板上停止。接着,在静止后,在大概5秒以内使注射针侧移动,通过将注射针从水滴分离,使水滴滚落,用高速相机以每5毫秒(200帧/秒)以静止画面拍摄水滴的行为。拍摄时的变焦倍率设为“w2”。仅水滴的前进侧的接触线在1秒移动了15~20mm时,视作滚落。将水滴的滚落时间(秒)作为横轴、将水滴的移动距离(mm)作为纵轴绘制成图,假定通过原点的一次函数,将通过最小二乘法拟合时的斜率作为滚落速度(mm/s)。
[0183]
实施例
[0184]
以下,通过实施例对本发明进行更具体的说明,但是本发明不受这些实施例的方式限定。
[0185]
(实施例1)
[0186]
在样品瓶中准备包含作为无机微粒的sylysia 310p(富士silysia化学制)27质量份、作为热固性树脂的zeffle gk-570(大金工业制)57质量份(固体成分换算)、作为固化剂的sumidur n-3300(住友covestro polyurethane制)13质量份、和作为拨水性成分a的unidyne tg-6501(大金工业制)3质量份的被膜形成用组合物。通过喷雾法在聚碳酸酯基材涂布上述被膜形成用组合物后,将该聚碳酸酯基材在130℃处理10分钟,由此形成被膜。
[0187]
(实施例2)
[0188]
作为无机微粒变更为13.5质量份的sylysia 310p和13.5质量份的sylophobic 100,除此以外,通过与实施例1同样的方法形成被膜。
[0189]
(实施例3)
[0190]
使用包含作为无机微粒的27质量份的平均粒径为2.7μm的sylysia 310p、作为热固性树脂的相当于上述聚合物f的主要由四氟乙烯和乙烯基单体构成的共聚物(以下,也有时称为聚合物f1。)57质量份(固体成分换算)、作为固化剂的13质量份的sumidur n-3300、和作为拨水性成分代替unidyne tg-6501的日本专利第5831599号的制备例2所述的共聚物(以下,也有时称为拨水性成分a。)3质量份的被膜形成用组合物,除此以外,通过与实施例1同样的方法形成被膜。
[0191]
(实施例4)
[0192]
使用包含作为无机微粒的13.5质量份的平均粒径为2.7μm的sylysia 310p和13.5质量份的平均粒径为2.7μm的sylophobic 100的混合物、作为热固性树脂的57质量份的上述聚合物f1(固体成分换算)、作为固化剂的13质量份的sumidur n-3300、作为拨水性成分代替unidynetg-6501的3质量份的拨水性成分a的被膜形成用组合物,除此以外,通过与实施例1同样的方法形成被膜。
[0193]
(实施例5)
[0194]
<拨水性成分b的制备>
[0195]
在四口烧瓶中,加入丙烯酸硬脂基酯(以下,记为sta。)35g(0.1mol)、二甲苯90g,进行30分钟氮气鼓气。其后,升温至90℃,加入0.085g的2,2’-偶氮二异丁腈(以下,有时也称为aibn。相对于丙烯酸硬脂基酯为0.5mol%),开始反应。反应开始2小时后,追加加入0.085g的aibn,使其反应4小时。在从最初的aibn投入6小时后,将反应溶液投入300ml的丙酮中,进行再沉淀操作,由此得到的沉淀物在130℃干燥2小时,由此,得到蜡性状的物质(拨水性成分b)。
[0196]
<涂膜的制备>
[0197]
作为拨水性成分,将unidyne tg-6501变更为拨水性成分b,除此以外,通过与实施例1同样的方法制备涂膜。
[0198]
(实施例6)
[0199]
<拨水性成分c的制备>
[0200]
在四口烧瓶中,加入sta29.42g(0.091mol)、甲基丙烯酸缩水甘油酯(以下,记为gma。)1.22g(0.009mol)、二甲苯90g,进行30分钟氮气鼓气。其后,升温至90℃,加入0.081g的aibn(相对于sta和gma的总量为0.5mol%),开始反应。反应开始2小时后,追加加入0.081g的aibn,使其反应4小时。在从最初的aibn投入的6小时后,将反应溶液投入300ml的甲醇中,进行再沉淀操作,由此得到的沉淀物在130℃干燥2小时,由此,得到蜡性状的物质
(拨水性成分c)。
[0201]
<涂膜的制备>
[0202]
作为拨水性成分,将unidyne tg-6501变更为拨水性成分c,除此以外,通过与实施例1同样的方法制备涂膜。
[0203]
(实施例7)
[0204]
<拨水性成分d的制备>
[0205]
将sta的投入量变更为19.6g(0.06mol),代替gma而变更为硬脂酸酰胺乙基丙烯酸酯11.03g(0.03mol),除此以外,通过与实施例6的拨液成分c的制备方法同样的方法,制备拨水性成分d。
[0206]
<涂膜的制作>
[0207]
作为拨水性成分,将unidyne tg-6501变更为拨水性成分d,除此以外,通过与实施例1同样的方法制备涂膜。
[0208]
(比较例1)
[0209]
作为无机微粒变更为45质量份的sylysia310p,作为热固性树脂变更为41质量份(固体成分换算)的zeffle gk-570,作为固化剂变更为9质量份的sumidur n-3300,作为拨水性成分变更为5质量份的unidyne tg-6501,除此以外,通过与实施例1同样的方法形成被膜。
[0210]
(比较例2)
[0211]
作为无机微粒变更为63质量份的sylysia310p,作为热固性树脂变更为24质量份(固体成分换算)的zeffle gk-570,作为固化剂变更为6质量份的sumidur n-3300,作为拨水性成分变更为7质量份的unidyne tg-6501,除此以外,通过与实施例1同样的方法形成被膜。
[0212]
(比较例3)
[0213]
作为无机微粒变更为13.5质量份的sylysia310p和13.5质量份的sylophobic 100的混合物,作为拨水性成分变更为全氟己基乙基四甲氧基硅烷(以下,有时也称为rf(c6)tms。)3质量份,除此以外,通过与实施例1同样的方法形成被膜。
[0214]
(比较例4)
[0215]
作为无机微粒变更为14质量份的sylysia310p和14质量份的sylophobic 100,作为拨水性成分变更为2质量份的rf(c6)tms,除此以外,通过与实施例1同样的方法形成被膜。
[0216]
(被膜的评价)
[0217]
<水的接触角(静态接触角)>
[0218]
水的接触角使用接触角计(协和界面科学公司“drop master 701”)。具体而言,使用水(2μl的液滴),对1个样品进行5点测定。静态接触角为150
°
以上时,该液体无法自立在基材表面存在,在该情况下,将注射器的针头作为支撑体测定静态接触角,将此时得到的值作为静态接触角。
[0219]
<铅笔硬度>
[0220]
铅笔硬度的测定方法根据jis k 5600-5进行。
[0221]
<72h浸水试验后的接触角>
[0222]
72h浸水试验后的接触角是将被膜在室温气氛下静置的10~30℃的水中浸渍72小时后,通过与上述的水的接触角的测定方法同样的测定方法来实施。此时的水接触角也通过接触角计(协和界面科学公司“drop master 701”)测定。
[0223]
<滚落角>
[0224]
滚落角的测定中,作为测定仪器,使用了drop master 701(协和界面科学株式会社制)。在注射针的前端形成20μl的水滴后,使载置于水平的试样载台上的涂敷基板表面与注射针前端的水滴的距离通过试样载台侧的可动而缓慢接近,在两者接触时使试样载台和注射针暂时静止,接着,使试样载台侧移动,缓慢分离,由此,使水滴在涂敷基板表面着滴。着滴后,在大概5秒以内使试样载台以2
°
/秒的倾斜速度倾斜,对倾斜角每1
°
以变焦倍率“w1”拍摄基板表面的水滴图像的静止画面。将水滴的后退侧的接触线开始移动时(移动0.1~1mm时)的试样载台的倾斜角度作为滚落角。
[0225]
<滚落速度>
[0226]
滚落速度的测定中,作为测定仪器,使用drop master 701(协和界面科学株式会社制)。在注射针的前端形成20μl的水滴后,使载置于预先倾斜30
°
的试样载台上的涂敷基板表面与注射针前端的水滴的距离通过试样载台侧的可动而缓慢接近,在两者接触时使试样载台和注射针暂时静止。在该时刻,水滴通过注射针在倾斜的涂敷基板上停止。接着,在静止后,在大概5秒以内使注射针侧移动,通过将注射针从水滴分离,使水滴滚落,用高速相机以每5毫秒(200帧/秒)以静止画面拍摄水滴的行为。拍摄时的变焦倍率设为“w2”。仅水滴的前进侧的接触线在1秒移动了15~20mm时,视作滚落。将水滴的滚落时间(秒)作为横轴、将水滴的移动距离(mm)作为纵轴绘制成图,假定通过原点的一次函数,将通过最小二乘法拟合时的斜率作为滚落速度(mm/s)。
[0227]
<耐磨耗性评价>
[0228]
摩擦试验
[0229]
耐磨耗性的评价通过以下记载的摩擦试验来实施。对于实施例1~5或比较例1、2中得到的各试验片,测定对水接触角,求出初期接触角。此后,对摩擦试验机(井元制作所制摩擦试验机“耐磨耗试验机(abrasion resistance tester)151e 3连规格”)的支架(与试样接触的面积:1cm 2
)作为擦拭纸安装无尘纸(商标名:kimwipe、日本制纸crecia制),以负荷100g/cm 2
涂擦一定次数。此后测定对水接触角,评价对于擦拭的耐磨耗性。这里的耐磨耗性能定义为能够维持超拨水状态(5次平均的静态接触角的值为150
°
以上、或者平均140
°
以上且加上其标准偏差为150
°
以上)的磨耗次数。在该试验中,接触角通过与<水的接触角(静态接触角)>同样的方法实施。
[0230][0231]
在表1和表2中,表示各实施例和比较例所得到的被膜的水的静态接触角(水20μl)、铅笔硬度、72小时浸水试验后的接触角、滚落角、滚落速度、耐磨耗性(仅表2)的结果。
[0232]
根据上述表所示的结果可知,各实施例中得到的被膜具有优异的超拨水性和优异的硬度和耐磨耗性,并且耐水性优异。另一方面,比较例1和2中,由于无机微粒的含量不合适,虽然具有超拨水性,但是出现硬度的降低。另外,比较例3中,不含式(1)所示的化合物的聚合物作为拨水性成分,因此,出现72小时浸水试验后的接触角的大幅降低。另外,比较例4中,也不含式(1)所示的化合物的聚合物作为拨水性成分,因此拨水性变差。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。