1.本发明公开了一种制备联苄化合物的方法,具体涉及一种可再生的生物质基苄醇及其苄醇类衍生物为底物,特别是利用生物质基的苄醇类化合物为底物,通过还原偶联过程,一步脱除羟基-碳碳偶联制备联苄化合物的方法。
背景技术:
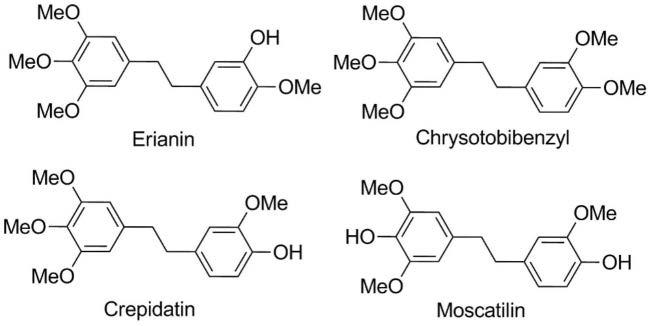
2.联苄化合物主要是具有1,2-二苯基乙烷母核骨架的芳香芪类化合物,虽然结构简单,但简单联苄大多是苯环和连接苯环的链桥上有简单的取代基,如甲基、甲氧基、羟基、氯等,或接有异戊烯单元和糖基等,而使其具有多样的结构类型,具有多种药理活性,特别是在保护心脏、保护神经、抗肿瘤、抗癌、抗炎等方面均表现出较好的活性,因此被广泛应用于医药行业。例如鼓槌石斛的提取物及从中分得的单体毛兰素(erianin)、玫瑰石斛素(crepidatin,4-羟基3,3’,4’,5
’‑
四甲氧基联苄)、鼓槌联苄(chrysotobibenzyl)和杓唇石斛素(moscatilin)等均有不同程度抗肿瘤活性(j.nat.prod.2007,70,24-28.)。其中毛兰素还对结肠癌、胃癌、肝癌和白血病等的癌细胞增殖有明显抑制作用。
[0003][0004]
3,4’,5-三羟基联苄(dihydroresveratrol,二氢白藜芦醇)作为白藜芦醇的同分异构体,常被用作抗肿瘤研究(nat.rev.drug discovery 2006,5,493-506.)。具有联苄结构的康普瑞汀(combretastatin)系列药物是一种天然血管靶向药物,具有极高的抗肿瘤活性,它通过利用肿瘤组织与正常组织内皮细胞的生理差异,可有选择性地抑制肿瘤微管蛋白的结合、改变其内皮细胞的骨架结构与形态,增强其血管渗透性、扰乱血流,从而引起肿瘤血管内皮细胞凋亡,导致次级肿瘤细胞死亡。康普瑞汀起效剂量低,使用时毒副作用较小,具有巨大的临床应用前景。
[0005][0006]
联苄化合物,特别是苯环上含有酚羟基、甲氧基等的天然联苄类化合物具有非常好的药理活性,是一类非常重要的药物或药物中间体。但是天然联苄化合物主要来源于苔藓植物、蕨类植物、被子植物,如兰科植物,百部科植物等。联苄化合物来源物种虽多,但植物资源分布于多种属,分布面积较广但密度小,种间杂生,不易区别和分离,难以大量采集。目前有关的人工合成方法较为少见。
[0007]
除此之外,联苄化合物也被广泛用于合成精细化学品的重要中间体,例如1,2-二苯基乙烷,是一种重要的精细化工中间体。上世纪90年代美国乙基公司利用二苯基乙烷作为关键性中间体成功开发出了新型阻燃剂十溴二苯乙烷,因其具有很好的光和热稳定性、强抗紫外线能力、毒性低、可回收循环使用等优点,可用于取代十溴二苯醚阻燃剂,特别适用于生产电脑、电话机、复印机、家电等的高档材料的阻燃。
[0008]
因此,联苄化合物是一种重要的药物或药物中间体,也是一种合成精细化学品不可替代的重要中间体,其合成在医药、阻燃、皮革加工等化工行业具有广泛的市场需求。
[0009]
目前对于联苄化合物的化学合成方法主要有三类:
[0010]
(1)以苯为主要原料与1,2-二卤代乙烷的傅-克烷基化反应;
[0011][0012]
该反应为放热反应,因此,降低反应温度有利于反应正想进行,但是温度过低反应速度太慢,反应周期长;温度高于75℃则反应速度过快副反应加剧,目标产物选择性下降。且该反应需要在严格无水条件下操作,反应过程中产生的氯化氢气体对设备有腐蚀作用,并且污染环境。
[0013]
(2)以氯化苄的偶联反应,主要有:
[0014]
2.1武兹偶联反应
[0015][0016]
此方法大量使用计量比金属钠,反应过程中生成的nacl附着在钠的表面,使其活性大大降低,并且金属钠的使用,操作条件需十分小心,否则极易发生爆炸或火灾,不适合工业化生产。
[0017]
2.2格氏偶联反应
[0018][0019]
此方法是氯化苄与计量比的金属镁首先在无水乙醚或thf中生成grignard试剂,再进行偶联反应得到目标产物,该方法大量使用乙醚,工业上难以保障安全性,并且grignard试剂的保存也是个问题,因此该法只适合实验室制备少量的产品。
[0020]
2.3氯化苄与活泼金属的偶联反应
[0021][0022]
以氯化苄为原料的偶联反应制备联苄化合物的方法的缺点是原料氯化苄的价格较高,并且催化剂与氯化苄的反应是计量反应,所需成本较高。
[0023]
(3)苯偶姻还原反应
[0024][0025]
该方法的原料和催化剂价格昂贵,难以实现工业化生产。
[0026]
综合上述,目前联苄化合物的合成路线中存在原料成本高,使用高危险、有毒的试剂和污染环境等问题。并且想要得到具有超高附加值的天然联苄药物,还需进行各种官能团化过程,路线复杂,成本更高。因此,开发一条相对廉价且高效的绿色制备联苄化合物的路径,对早日实现联苄化合物的工业化生产具有重要的意义。
技术实现要素:
[0027]
本发明的目的在于提供一种利用可再生生物质基苄醇以及苄醇类衍生物,特别是利用生物质基的苄醇类化合物为底物,通过还原偶联过程,一步脱除羟基-碳碳偶联制备联苄化合物的新路径。
[0028]
本发明的技术方案时,一种制备联苄化合物的方法,以生物质基苄醇或苄醇类衍生物的为底物,在溶剂中与还原剂混合、与催化剂接触,经还原偶联反应,一步脱羟基-碳碳偶联制备联苄化合物的方法;
[0029]
所述苄醇类衍生物为:苯甲醇,对氟苯甲醇,对氯苯甲醇,对溴苯甲醇,对甲基苯甲醇,二苯甲醇,三苯基甲醇,3,4-二甲基苯甲醇,3,5-二甲基苯甲醇,3,4,5-三甲基苯甲醇,9-芴醇,苏和香醇,对羟基苯甲醇,大茴香醇,藜芦醇,香草醇,3,5-二甲氧基苯甲醇,3,4,5-三甲氧基苯甲醇,4-羟基-3,5-二甲氧基苯甲醇,3,4-二羟基苯甲醇,松柏醇,芥子醇,对香豆醇中的至少一种。
[0030]
所述底物初始反应摩尔浓度为0.1-10mol/l。
[0031]
优选所述制备联苄化合物的反应方程式为:
[0032][0033]
优选所述还原偶联反应的催化剂的活性成分选自有机金属配合物或其衍生物;金属元素选自镍、铜、钯、铑、铂或铼;
[0034]
优选所述还原偶联反应的催化剂还包括载体,所述载体选自二硫化钼,三氧化钼,三氧化钨,五氧化二铌,二氧化钛,二氧化硅,二氧化锌,二氧化锆,活性炭,石墨烯;
[0035]
优选所述催化剂的用量与底物的摩尔比为0.01-2。
[0036]
优选所述还原剂为亚磷酸三乙酯,二氢吲哚,异丙醇、正丁醇、3-戊醇、2-辛醇等醇类,碳粉,亚硫酸钠等其中的一种或两种以上;
[0037]
优选所述还原剂的用量与底物的摩尔比为0.5~5。
[0038]
优选所述溶剂为甲醇、乙醇、异丙醇、正丁醇、叔丁醇等醇类和乙腈,1,4-二氧六环,二氯甲烷,二氯乙烷,四氢呋喃等的一种或者多种。
[0039]
优选所述反应的温度为30~350℃,反应时间为不少于1h;优选反应温度为25~280℃,优选反应时间为1~72h,更优1~24;
[0040]
优选所述反应气氛可以为氩气或氮气或空气中的一种或两种。
[0041]
优选所述反应底物的转化率≥60%,产物的收率≥50%。
[0042]
本发明的有益效果
[0043]
(1)利用苄醇以及苄醇类衍生物,特别是利用生物质基的苄醇类化合物,尤其是木质素衍生的苄醇类化合物为底物,底物本身具有特殊的酚羟基,甲氧基等官能团,不涉及多步且复杂的官能团化过程就可以制备多种天然联苄药物。
[0044]
(2)反应过程操作简单,相对绿色,避免了使用高危险、强腐蚀性的有害原料。
[0045]
(3)根据需要,利用含有不同官能团的底物,可以制备的联苄产物种类繁多,满足不同的需求。
具体实施方式
[0046]
本发明公开了一种全新的反应路径以一种苄醇及其苄醇类衍生物为底物,特别是利用生物质基的苄醇类化合物为底物,以镍,铜,钯,铑或铂等为催化剂,通过还原偶联过程,一步脱除羟基-碳碳偶联制备联苄化合物的方法。本领域研究和技术人员可以借鉴本文内的内容,适当进行改进。特别需要指出的是,所有类似的替换和改动对本领域研究和技术人员来说是显而易见的,它们都被视为包括在本发明中。本发明的方法以及应用已经通过较佳的实例进行描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明的技术。
[0047]
底物苄醇类衍生物包含:苯甲醇,对氟苯甲醇,对氯苯甲醇,对溴苯甲醇,对甲基苯甲醇,二苯甲醇,三苯基甲醇,3,4-二甲基苯甲醇,3,5-二甲基苯甲醇,3,4,5-三甲基苯甲醇,9-芴醇,苏和香醇(1-苯乙醇),对羟基苯甲醇,大茴香醇(对甲氧基苯甲醇),藜芦醇(3,
4-二甲氧基苯甲醇),香草醇(4-羟基-3-甲氧基苯甲醇),3,5-二甲氧基苯甲醇,3,4,5-三甲氧基苯甲醇,4-羟基-3,5-二甲氧基苯甲醇,3,4-二羟基苯甲醇,松柏醇(4-羟基-3-甲氧基肉桂醇),芥子醇(4-羟基-3,5-二甲氧基肉桂醇),对香豆醇(4-羟基肉桂醇)中的一种或两种以上。
[0048]
所述催化剂包含镍、铜、钯、铑、铂或铼等有机金属配合物及其衍生物等的一种或者多种,也可将镍、铜、钯、铑、铂或铼等一种或者多种负载在二硫化钼,三氧化钼,三氧化钨,五氧化二铌,二氧化钛,二氧化硅,二氧化锌,二氧化锆,活性炭,石墨烯等载体上的多相催化剂。所述催化剂的用量为原料的1~20mol%,优选用量为0.5~15mol%,最优选用量为3~10mol%。
[0049]
所述还原剂可以为亚磷酸三乙酯,二氢吲哚,异丙醇、正丁醇、3-戊醇、2-辛醇等醇类,碳粉,亚硫酸钠等其中的一种或两种以上。
[0050]
所述还原剂的用量与底物的摩尔比为0.5-5,优选用量摩尔比为0.5~3,最优选用量摩尔比为1~2。
[0051]
反应温度为30~350℃,优选反应温度为25~280℃。
[0052]
反应时间为1~72h,在一定时间范围内,随反应时间增加转化率提高,但是反应时间延长到一定时间以后,转化率和产物选择性稳定,优选反应时间为1~24h。
[0053]
实施例1
[0054]
在反应釜中,分别加入1mmol二苯基甲醇,0.01mmol reo
x
/ceo2,1mmol亚磷酸三乙酯,20ml1,4-二氧六环,用氮气置换,在120℃下反应18h,反应结束后,降至室温。气相内标法定量分析,其中底物转化率》99%,产物四苯基乙烷的收率为93%。抽滤,回收催化剂。
[0055]
实施例2
[0056]
在反应釜中,分别加入1mmol二苯基甲醇,0.01mmolreo
x
/tio2,20ml异丙醇,用氩气置换,在160℃下反应18h,反应结束后,降至室温。气相内标法定量分析,底物完全转化,且联苄产物四苯基乙烷的收率95%。抽滤,回收催化剂。
[0057]
实施例3
[0058]
在反应釜中,分别加入1mmol二苯基甲醇,0.05mmol乙酰丙酮镍,1mmol三苯基膦,20ml正丁醇,用氮气置换,在200℃下反应14h,反应结束后,降至室温。反应液气相内标法定量分析,底物转化率93%,且联苄产物四苯基乙烷的收率78%。
[0059]
实施例4
[0060]
在反应釜中,分别加入1mmol苏合香醇(1-苯乙醇),0.01mmol 8-羟基喹啉铜,1.5mmol亚磷酸三乙酯,20ml1,4-二氧六环,用氩气置换,在120℃下反应18h,反应结束后,降至室温。气相内标法定量,底物转化率为86%,联苄产物2,3-二苯基丁烷的收率为73%。
[0061]
实施例5
[0062]
在反应釜中,分别加入1mmol大茴香醇,0.03mmol 8-羟基喹啉镍,1mmol双环咪唑啉,20ml正丁醇,用空气置换,在150℃下反应18h,反应结束后,降至室温。用气相内标法定量,底物完全转化,联苄产物4,4
’‑
二甲氧基联苄的收率为82%。
[0063]
实施例6
[0064]
在反应釜中,分别加入1mmol大茴香醇,0.1mmol 8-羟基喹啉氧钼,1mmol三苯基膦,20ml1,4-二氧六环,用氩气置换,在220℃下反应8h,反应结束后,降至室温。气相内标法
定量,底物完全转化,联苄产物4,4
’‑
二甲氧基联苄的收率为60%。
[0065]
实施例7
[0066]
在反应釜中,分别加入1mmol大茴香醇,0.1mmol醋酸钯,1mmol碳粉,20ml四氢呋喃,用氮气置换,在160℃下反应8h,反应结束后,降至室温。气相内标法定量,底物完全转化,联苄产物4,4
’‑
二甲氧基联苄的收率为74%。
[0067]
实施例8
[0068]
在反应釜中,分别加入1mmol大茴香醇,0.1mmol乙酰丙酮镍,1mmol希夫碱,1mmol亚磷酸三乙酯,20ml1,4-二氧六环,用氩气置换,在100℃下反应18h,反应结束后,降至室温。气相内标法定量,底物转完全转化,联苄产物4,4
’‑
二甲氧基联苄的收率为76%。
[0069]
实施例9
[0070]
在反应釜中,分别加入1mmol藜芦醇,0.03mmolre2o7,1mmol三苯基膦,20ml四氢呋喃,用氮气置换,在150℃下反应18h,反应结束后,降至室温。气相内标法定量,底物完全转化,且联苄产物3,3’,4,4
’‑
四甲氧基联苄的收率为78%。
[0071]
实施例10
[0072]
在反应釜中,分别加入1mmol藜芦醇,0.1mmol三氧化钼,1mmol 8-羟基喹啉,1mmol三苯基膦,20ml1,4-二氧六环,用氮气和氩气(体积比1:1)置换,在220℃下反应8h,反应结束后,降至室温。气相内标法定量,底物完全转化,且联苄产物3,3’,4,4
’‑
四甲氧基联苄的收率为70%。
[0073]
实施例11
[0074]
在反应釜中,分别加入1mmol香草醇,0.05mmolch3reo3,1mmol三苯基膦,20ml1,4-二氧六环,用空气置换,在180℃下反应10h,反应结束后,降至室温。气相内标法定量,底物完全转化,联苄产物4,4
’‑
二羟基3,3
’‑
二甲氧基联苄的收率为88%。
[0075]
实施例12
[0076]
在反应釜中,分别加入1mmol香草醇,0.1mmol三氧化钼,0.3mmol 8-羟基喹啉,1mmol三苯基膦,20ml1,4-二氧六环,用氮气置换,在200℃下反应19h,反应结束后,降至室温。气相内标法定量,底物完全转化,联苄产物4,4
’‑
二羟基3,3
’‑
二甲氧基联苄的收率为93%。
[0077]
实施例13
[0078]
在反应釜中,分别加入1mmol 9-羟基芴醇,0.03mmol re2o7,1.5mmol三苯基膦,20ml1,4-二氧六环,用氮气置换,在140℃下反应16h,反应结束后,降至室温。气相定量分析,底物完全转化,产物收率为98%。
[0079]
实施例14
[0080]
在反应釜中,分别加入1mmol 9-羟基芴醇,0.1mmol氯化铜,1mmol三苯基膦,20ml1,4-二氧六环,用氮气置换,在160℃下反应14h,反应结束后,降至室温。气相定量,底物转化率为60%,联苄产物收率53%。
[0081]
实施例15
[0082]
在反应釜中,分别加入0.5mmol 3,4,5-三甲氧基苯甲醇和0.5mmol香草醇,0.1mmol8-羟基喹啉氧钼,1mmol三苯基膦,20ml1,4-二氧六环,用氮气置换,在220℃下反应10h,反应结束后,降至室温。气相内标法定量,底物均完全转化。联苄产物包含3,3’,4,4’,
5,5
’‑
六甲氧基联苄、4,4
’‑
二羟基3,3
’‑
二甲氧基联苄和玫瑰石斛素(crepidatin,4-羟基3,3’,4’,5
’‑
四甲氧基联苄),且总收率为53%。
[0083]
实施例16
[0084]
在反应釜中,分别加入0.5mmol 4-羟基3,5-二甲氧基苯甲醇和0.5mmol香草醇,0.1mmol8-羟基喹啉氧钼,20ml正丁醇,用氮气置换,在220℃下反应10h,反应结束后,降至室温。气相内标法定量,两种底物都完全转化,且联苄产物包含4,4
’‑
二羟基3,3
’‑
二甲氧基联苄、4,4
’‑
二羟基3,3’,5,5
’‑
四甲氧基联苄和毛兰素素(erianin,4,4
’‑
二羟基3,3’,5
’‑
三甲氧基联苄),且总收率为67%。
[0085]
实施例17
[0086]
在反应釜中,分别加入1mmol二苯基甲醇,0.1mmol pd-pt/石墨烯,1.2mmol 2,4,6-三甲基吡啶,2mmol锌粉,3ml thf,用空气置换,在90℃下反应15h,反应结束后,降至室温。气相内标法定量,底物转化率为85%,联苄产物的收率为79%。
[0087]
以上所述,仅为本发明部分的优选具体实施例,但是本发明的保护范围并不仅限于此,也不因各实施例的先后次序对本发明造成任何限制,任何熟悉本发明技术领域的技术人员在本发明报道的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围不仅限于以上实施例,应该以权利要求的保护范围为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。