手性nhc催化制备手性3,2`-吡咯烷螺环吲哚骨架化合物的方法
技术领域
1.本发明属于有机合成技术领域,具体涉及手性nhc催化制备手性3,2
′‑
吡咯烷螺环吲哚骨架化合物的方法。
背景技术:
2.3,2
′‑
吡咯烷螺环吲哚骨架化合物具有特别的空间结构且广泛存在于天然生物碱与许多生物活性分子中,研究显示该类化合物具有很好的生物活性,因而在有机化学、天然药物化学以及药物的研究中具有十分重要的意义。目前报道的催化合成3,2
′‑
吡咯烷螺环吲哚骨架类化合物的方法可分为有机金属催化、非金属催化以及协同催化方法。金属催化方法主要包括了铜、钯、铑和汞等金属作为催化剂;有机非金属催化主要包括了手性金鸡纳生物碱衍生的硫脲催化剂、奎宁衍生的催化剂、手性磷酸、二乙胺和四氯化锡等;协同反应方法主要包含了有机金属与非金属有机试剂共同作用催化底物合成最终产物。但采用上述方法存在技术路线复杂,时间长,成本高的问题。
3.氮杂环卡宾(nhc)作为一类重要的仿生有机催化剂,通过将醛类化合物的反应极性发生翻转,使其由传统的亲电性转化为亲核性。氮杂环卡宾催化的各类反应为碳-碳键的构筑及各类复杂天然产物的合成提供了新的合成方法和策略。但现有技术中,尚未有采用氮杂环卡宾催化剂制备3,2
′‑
吡咯烷螺环吲哚骨架化合物的报道。因此亟需提供一种氮杂环卡宾催化制备手性3,2
′‑
吡咯烷螺环吲哚骨架化合物的方法,解决现有技术中手性3,2
′‑
吡咯烷螺环吲哚骨架化合物合成中技术路线复杂,时间长,成本高的问题。
技术实现要素:
4.本发明的目的在于,提供手性nhc催化制备手性3,2
′‑
吡咯烷螺环吲哚骨架化合物的方法,反应条件温和,路线短,成本低。
5.为实现上述目的,本发明采用的技术方案为:
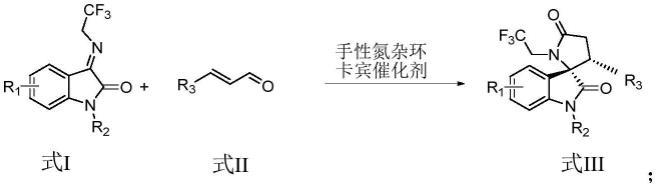
6.本发明提供的一种手性nhc催化制备手性3,2
′‑
吡咯烷螺环吲哚骨架化合物的方法,式i化合物与式ii化合物在手性氮杂环卡宾催化剂的作用下,反应生成式iii所示的手性3,2
′‑
吡咯烷螺环吲哚骨架化合物;其反应式为:
[0007][0008]
其中,r1为卤原子、甲基、甲氧基、硝基、4,6-二氟或5,6-二氟;
[0009]
r2为甲基、取代或未取代的苯基、萘基、茚基、呋喃基、嘧啶基、吲哚基、噻唑基或噻吩基;
[0010]
r3为取代或未取代的苯基、萘基、茚基、呋喃基、嘧啶基、吲哚基、噻唑基或噻吩基。
[0011]
本发明的部分实施方案中,所述手性氮杂环卡宾催化剂的结构式如下所示:
[0012][0013]
本发明的手性nhc催化制备手性3,2
′‑
吡咯烷螺环吲哚骨架化合物的方法的反应过程如下:
[0014][0015]
式ii化合物与手性氮杂环卡宾催化剂反应,得到homoenolates中间体ⅰ, homoenolates中间体ⅰ与亲电体式i化合物进行[2 3]环化反应,氮杂环卡宾催化剂离去继续参与循环过程,不断催化生成式ⅲ所示的螺环化合物。
[0016]
本发明的部分实施方案中,式i化合物与式ii化合物的摩尔比为0.8~1.2: 0.8~1.2,优选为1:1.2。
[0017]
本发明的部分实施方案中,式i化合物与手性氮杂环卡宾催化剂的摩尔比为 4~6:1,优选为4~5:1。
[0018]
本发明的部分实施方案中,所述方法包括以下步骤:
[0019]
s1.保护气体条件下,将手性氮杂环卡宾催化剂与碱于溶剂中反应,优选地,所述碱包括三乙胺、dipea、碳酸钾、碳酸氢钾中的至少一种;
[0020]
s2.加入式i化合物、式ii化合物,反应生成式iii所示的手性3,2
′‑
吡咯烷螺环吲哚骨架化合物。
[0021]
本发明中s1中,通过将催化剂和碱反应一段时间,以活化催化剂。本发明的部分实
施方案中,s1中,手性氮杂环卡宾催化剂与三乙胺的摩尔比为1:10~15。
[0022]
本发明的部分实施方案中,s1中所述的溶剂包括二氯甲烷、乙腈、2-甲基四氢呋喃、1,2-二氯乙烷和甲苯。
[0023]
本发明的部分实施方案中,s1中,在低温条件下反应,优选为0~4℃条件下反应,更优选为0℃条件下反应。
[0024]
本发明的部分实施方案中,s2中,在低温条件下反应,优选为0~4℃条件下反应,更优选为0℃条件下反应。
[0025]
本发明的部分实施方案中,s1中反应时间为10~60min,优选为30min。
[0026]
本发明的一个实施例中,手性nhc催化制备手性3,2
′‑
吡咯烷螺环吲哚骨架化合物的方法的反应式为:
[0027][0028]
其反应过程为:
[0029]
(1)1.2摩尔当量的肉桂醛与0.2摩尔当量手性氮杂环卡宾催化剂反应,得到homoenolates中间体ⅰ,反应温度为0℃,反应溶剂为二氯甲烷;
[0030]
(2)homoenolates中间体ⅰ与1.0摩尔当量亲电体1-甲基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮进行[2 3]环化反应,氮杂环卡宾催化剂离去继续参与循环过程,不断催化生成螺环化合物(3s,3
′
r)-1-甲基-3
′‑
苯基-1
′‑
(2,2,2-三氟乙基)螺 [二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮衍生物,反应温度为0℃,反应溶剂为二氯甲烷。
[0031]
本发明中所述的保护气体包括氮气、惰性气体。
[0032]
本发明中英文缩写对应的中文名称为:
[0033]
et3n:三乙胺
[0034]
dcm:二氯甲烷dipea:n,n-二异丙基乙胺
[0035]
与现有技术相比,本发明具有以下有益效果:
[0036]
具有简单结构单元的反应物分子取代3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚
ꢀ‑
2-酮和醛类化合物能有效在氮杂环卡宾的催化作用下,高效的制备了(3s,3
′
r)-3
′‑ꢀ
苯基-1
′‑
(2,2,2-三氟乙基)螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮衍生物,并具有衍生物普适性好、优异的产率和高对映选择性等优点。
附图说明
[0037]
附图1为实施例5制得的
ⅲ‑
5化合物的氢谱图;
[0038]
附图2为实施例5制得的
ⅲ‑
5化合物的碳谱图;
[0039]
附图3为实施例5制得的
ⅲ‑
5化合物的非手性液相图;
[0040]
附图4为实施例5制得的
ⅲ‑
5化合物的手性液相图。
[0041]
附图5为实施例5制得的
ⅲ‑
5化合物的质谱图。
具体实施方式
[0042]
联系如下实施例,将更好地理解本发明的优点和制备过程,这些实施例旨在阐述而不是限制本发明的范围。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0043]
本发明实施例中所用的氮杂环卡宾3e的结构式为:
[0044][0045]
实施例1
[0046]
本实施例公开了化合物
ⅲ‑
1:(3s,3
′
r)-5-溴-1-甲基-3
′‑
苯基-1
′‑
(2,2,2-三氟乙基)螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮的合成方法,具体为:
[0047][0048]
在n2保护下,向干燥的10ml反应管中分别加入11.85mg氮杂环卡宾3e(0.03mmol)和39.53mget3n(0.39mmol),再加入1.0ml二氯甲烷,在0℃下反应30分钟,再加入50mg5-溴-1-甲基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮(0.16mmol)和24.78mg肉桂醛(0.19mmol)的二氯甲烷溶液(3ml),继续在0℃下反应,tlc监测反应完全后,浓缩溶剂,所得粗产品通过柱层析分离,洗脱机极性石油醚:乙酸乙酯=5:1得目标产物
ⅲ‑
1,收率为85%,对映体比例为96:4。
[0049]1hnmr(600mhz,cdcl3)δ7.55
–
7.50(m,2h),7.24
–
7.18(m,1h),7.18
–
7.11(m,2h),6.88(dd,j=8.2,1.3hz,2h),6.55(d,j=8.8hz,1h),4.01(dq,j=15.3,9.5hz,1h),3.78(dd,j=13.1,7.9hz,1h),3.58(dd,j=16.4,13.2hz,1h),3.52(dt,j=15.2,8.7hz,1h),2.75(dd,j=16.3,7.9hz,1h),2.70(s,3h).
13
cnmr(151mhz,cdcl3)δ175.72,172.91,143.51,133.82,132.47,128.43,128.26,127.84,127.26,126.19,123.19(q,j=280.3hz),115.74,110.20,73.50,51.82,42.44(q,j=35.5hz),32.79,25.75.hrms(esi)calculated[m na]
forc
20h16
brf3n2o2na:475.0245,found:475.0239.
[0050]
手性分析通过hplc,具体条件为:ib柱,n-hexane/ethanol/diethylamine=80/20/0.1,1.0ml/min,λ=254nm。
[0051]
实施例2
[0052]
本实施例公开了化合物
ⅲ‑
2:(3s,3
′
r)-5-氯-1-甲基-3
′‑
苯基-1
′‑
(2,2,2-三氟乙基)螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮的合成方法,具体为:
[0053][0054]
在n2保护下,向干燥的10ml反应管中分别加入13.74mg氮杂环卡宾3e (0.04mmol)和45.82mg et3n(0.45mmol),再加入1.5ml二氯甲烷,在0℃下反应30分钟,再加入50mg 5-氯-1-甲基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮 (0.18mmol)和28.73mg肉桂醛(0.22mmol)的二氯甲烷溶液(3ml),继续在0℃下反应,tlc监测反应完全后,浓缩溶剂,所得粗产品通过柱层析分离,洗脱机极性石油醚:乙酸乙酯=4:1得目标产物
ⅲ‑
2,收率为83%,对映体比例为95:5。
[0055]1h nmr(600mhz,cdcl3)δ7.40
–
7.34(m,2h),7.24
–
7.18(m,1h),7.18
–ꢀ
7.12(m,2h),6.90
–
6.85(m,2h),6.59(d,j=8.2hz,1h),3.99(dq,j=15.4,9.5hz, 1h),3.78(dd,j=13.1,7.8hz,1h),3.62
–
3.48(m,2h),2.75(ddd,j=16.4,7.9,0.8 hz,1h),2.70(s,3h).
13
c nmr(151mhz,cdcl3)δ175.76,173.03,143.01,132.48, 130.92,128.69,128.42,128.26,127.84,125.85,124.53,123.19(q,j=280.3hz), 109.75,73.59,51.81,42.46(q,j=35.7hz),32.80,25.78.hrms(esi)calculated [m na]
for c
20h16
clf3n2o2na:431.0750,found:431.0744.
[0056]
手性分析通过hplc,具体条件为:ic柱,n-hexane/2-propanol=80/20,0.8 ml/min,λ=254nm。
[0057]
实施例3
[0058]
本实施例公开了化合物
ⅲ‑
3:(3s,3
′
r)-5-氟-1-甲基-3
′‑
苯基-1
′‑
(2,2,2-三氟乙基) 螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮的合成方法,具体为:
[0059][0060]
在n2保护下,向干燥的10ml反应管中分别加入14.58mg氮杂环卡宾3e (0.04mmol)和48.64mg et3n(0.48mmol),再加入1.5ml二氯甲烷,在0℃下反应30分钟,再加入50mg 5-氟-1-甲基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮 (0.19mmol)和30.49mg肉桂醛(0.23mmol)的二氯甲烷溶液(3ml),继续在0℃下反应,tlc监测反应完全后,浓缩溶剂,所得粗产品通过柱层析分离,洗脱机极性石油醚:乙酸乙酯=4:1得目标产物
ⅲ‑
3,收率为87%,对映体比例为90:10。
[0061]1h nmr(600mhz,cdcl3)δ7.25
–
7.07(m,5h),6.91
–
6.83(m,2h),6.60 (dd,j=8.5,4.0hz,1h),3.90(dq,j=15.4,9.4hz,1h),3.77(dd,j=13.1,7.8hz, 1h),3.65
–
3.54(m,
2h),2.76(ddd,j=16.4,7.9,0.7hz,1h),2.72(s,3h).
13
cnmr(151mhz,cdcl3)δ175.89,173.21,159.36(d,j=243.2hz),140.41(d,j=2.2hz),132.56,128.39,128.24,127.85,125.73(d,j=7.7hz),123.23(q,j=280.3hz),117.39(d,j=23.3hz),112.30(d,j=25.1hz),109.49(d,j=7.9hz),73.89,51.74,42.60(q,j=35.6hz),32.87,25.81.hrms(esi)calculated[m na]
forc
20h16
f4n2o2na:415.1046,found:415.1040.
[0062]
手性分析通过hplc,具体条件为:ic柱,n-hexane/2-propanol=80/20,0.8ml/min,λ=254nm。
[0063]
实施例4
[0064]
本实施例公开了化合物
ⅲ‑
4:(3s,3
′
r)-7-溴-1-甲基-3
′‑
苯基-1
′‑
(2,2,2-三氟乙基)螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮的合成方法,具体为:
[0065][0066]
在n2保护下,向干燥的10ml反应管中分别加入11.85mg氮杂环卡宾3e(0.03mmol)和39.53mget3n(0.39mmol),再加入1.0ml二氯甲烷,在0℃下反应30分钟,再加入50mg7-溴-1-甲基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮(0.16mmol)和24.78mg肉桂醛(0.19mmol)的二氯甲烷溶液(3ml),继续在0℃下反应,tlc监测反应完全后,浓缩溶剂,所得粗产品通过柱层析分离,洗脱机极性石油醚:乙酸乙酯=5:1得目标产物
ⅲ‑
4,收率为80%,对映体比例为95:5。
[0067]1hnmr(600mhz,cdcl3)δ7.51(dd,j=8.1,1.1hz,1h),7.32(dd,j=7.4,1.1hz,1h),7.26
–
7.22(m,1h),7.18(t,j=7.6hz,2h),7.09(dd,j=8.2,7.3hz,1h),6.83(dd,j=7.3,1.7hz,2h),3.94(dq,j=15.3,9.4hz,1h),3.74(dd,j=13.1,7.8hz,1h),3.62
–
3.47(m,2h),3.07(s,3h),2.76(dd,j=16.4,7.8hz,1h).
13
cnmr(151mhz,cdcl3)δ175.90,173.93,141.86,136.46,132.41,128.54,128.28,127.73,127.37,124.31,123.22,123.21(q,j=280.9hz),103.08,73.28,52.24,42.45(q,j=35.8hz),32.78,29.25.hrms(esi)calculated[m na]
forc
20h16
brf3n2o2na:475.0245,found:475.0239.
[0068]
手性分析通过hplc,具体条件为:ib柱,n-hexane/ethanol/diethylamine=80/20/0.1,1.0ml/min,λ=254nm。
[0069]
实施例5
[0070]
本实施例公开了化合物
ⅲ‑
5:(3s,3
′
r)-1-苄基-3
′‑
苯基-1
′‑
(2,2,2-三氟乙基)螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮的合成方法,具体为:
[0071][0072]
在n2保护下,向干燥的10ml反应管中分别加入11.92mg氮杂环卡宾3e (0.03mmol)和39.76mg et3n(0.39mmol),再加入1.0ml二氯甲烷,在0℃下反应30分钟,再加入50mg 1-苄基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮(0.16 mmol)和24.93mg肉桂醛(0.19mmol)的二氯甲烷溶液(3ml),继续在0℃下反应, tlc监测反应完全后,浓缩溶剂,所得粗产品通过柱层析分离,洗脱机极性石油醚:乙酸乙酯=3:1得目标产物
ⅲ‑
5,收率为89%,对映体比例为98:2。
[0073]1h nmr(600mhz,cdcl3)δ7.41(dd,j=7.2hz,1h),7.31(t,j=7.4hz,1h), 7.26
–
7.22(m,1h),7.23
–
7.12(m,4h),7.08(t,j=7.6hz,2h),6.96(d,j=7.2hz, 2h),6.45(d,j=7.8hz,1h),6.38(d,j=7.6hz,2h),4.95(d,j=16.0hz,1h),4.17 (d,j=16.0hz,1h),4.05(dq,j=15.3,9.6hz,1h),3.92(dd,j=13.3,7.8hz,1h), 3.71(dd,j=16.3,13.3hz,1h),3.52
–
3.42(m,1h),2.79(dd,j=16.3,7.8hz,1h). 13
c nmr(151mhz,cdcl3)δ175.93,173.68,143.99,134.49,133.07,130.98, 128.64,128.62,128.42,128.34,127.28,126.38,124.26,123.84,123.30,123.27(q,j =280.5hz),110.20,73.22,51.39,43.81,42.24(q,j=35.7hz),33.19.hrms (esi)calculated[m na]
for c
26h21
f3n2o2na:473.1453,found:473.1449.
[0074]
手性分析通过hplc,具体条件为:ib柱,n-hexane/ethanol/diethylamine =80/20/0.1,1.0ml/min,λ=254nm。
[0075]
实施例6
[0076]
本实施例公开了化合物
ⅲ‑
6:(3s,3
′
r)-1-苄基-3
′‑
(4-溴苯基)-1
′‑
(2,2,2-三氟乙基)螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮的合成方法,具体为:
[0077][0078]
在n2保护下,向干燥的10ml反应管中分别加入11.92mg氮杂环卡宾3e (0.03mmol)和39.76mg et3n(0.39mmol),再加入1.0ml二氯甲烷,在0℃下反应30分钟,再加入50mg 1-苄基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮(0.16 mmol)和39.81mg对溴肉桂醛(0.19mmol)的二氯甲烷溶液(3ml),继续在0℃下反应,tlc监测反应完全后,浓缩溶剂,所得粗产品通过柱层析分离,洗脱机极性石油醚:乙酸乙酯=5:1得目标产物
ⅲ‑
6,收率为85%,对映体比例为92:8。
[0079]1hnmr(600mhz,cdcl3)δ7.40(dd,j=7.3hz,1h),7.31
–
7.27(m,3h),7.24
–
7.15(m,4h),6.82(d,j=8.5hz,2h),6.52(d,j=7.9hz,1h),6.48
–
6.43(m,2h),5.02(d,j=16.0hz,1h),4.16(d,j=15.9hz,1h),4.05(dq,j=15.4,9.6hz,1h),3.86(dd,j=13.2,7.8hz,1h),3.64(dd,j=16.3,13.2hz,1h),3.45(dq,j=15.3,8.6hz,1h),2.78(dd,j=16.2,7.8hz,1h).
13
cnmr(151mhz,cdcl3)δ175.47,173.52,144.00,134.36,132.20,131.75,131.19,130.09,128.78,127.57,126.41,124.23,123.51,123.44,123.22(q,j=279.4hz),122.57,110.29,72.85,50.73,43.96,42.22(q,j=36.2hz),33.15.hrms(esi)calculated[m na]
forc
26h20
brf3n2o2na:551.0558,found:551.0551.
[0080]
手性分析通过hplc,具体条件为:ib柱,n-hexane/ethanol/diethylamine=80/20/0.1,1.0ml/min,λ=254nm。
[0081]
实施例7
[0082]
本实施例公开了化合物
ⅲ‑
7:(3s,3
′
r)-1-苄基-3
′‑
(4-溴苯基)-1
′‑
(2,2,2-三氟乙基)螺[二氢吲哚-3,2
′‑
吡咯烷]-2,5
′‑
二酮的合成方法,具体为:
[0083][0084]
在n2保护下,向干燥的10ml反应管中分别加入11.92mg氮杂环卡宾3e(0.03mmol)和39.76mget3n(0.39mmol),再加入1.0ml二氯甲烷,在0℃下反应30分钟,再加入50mg1-苄基-3-((2,2,2-三氟乙基)亚氨基)-1h-吲哚-2-酮(0.16mmol)和31.42mg对氯肉桂醛(0.19mmol)的二氯甲烷溶液(3ml),继续在0℃下反应,tlc监测反应完全后,浓缩溶剂,所得粗产品通过柱层析分离,洗脱机极性石油醚:乙酸乙酯=5:1得目标产物
ⅲ‑
7,收率为81%,对映体比例为90:10。
[0085]1hnmr(600mhz,cdcl3)δ7.40(dd,j=6.8hz,1h),7.30
–
7.26(m,1h),7.23
–
7.17(m,4h),7.16
–
7.12(m,2h),6.90
–
6.85(m,2h),6.52(d,j=7.8hz,1h),6.48
–
6.41(m,2h),5.01(d,j=15.9hz,1h),4.16(d,j=15.9hz,1h),4.05(dq,j=15.4,9.6hz,1h),3.88(dd,j=13.2,7.8hz,1h),3.65(dd,j=16.3,13.2hz,1h),3.45(dq,j=15.3,8.6hz,1h),2.78(dd,j=16.2,7.8hz,1h).
13
cnmr(151mhz,cdcl3)δ175.49,173.51,144.00,134.39,134.37,131.66,131.18,129.76,128.80,128.69,127.56,126.41,124.23,123.53,123.44,123.22(q,j=280.9hz),110.26,72.94,50.68,43.93,42.23(q,j=36.2hz),33.18.hrms(esi)calculated[m na]
forc
26h20
clf3n2o2na:507.1063,found:507.1051.
[0086]
手性分析通过hplc,具体条件为:ib柱,n-hexane/ethanol/diethylamine=80/20/0.1,1.0ml/min,λ=254nm。
[0087]
以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的
所有其他实施例,都属于本发明保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。