the treatment of patients with cancer.cancer j sci am.2000feb;6suppl 1:s2-7.
9.4.bassi p.bcg(bacillus of calmette guerin)therapy of high-risk superficial bladder cancer.surg oncol.2002jun;11(1-2):77-83.
10.5.antonia s,et al.current developments of immunotherapy in the clinic.curr opin immunol.2004apr;16(2):130-6.
11.6.gu ss,et al.therapeutically increasing mhc-i expression potentiates immune checkpoint blockade.cancer discov.2021jun;11(6):1524-1541.
12.7.yang l,et al.tgf-beta and immune cells:an important regulatory axis in the tumor microenvironment and progression.trends immunol.2010jun;31(6):220-7.
13.8.dong h,et al.b7-h1,a third member of the b7 family,co-stimulates t-cell proliferation and interleukin-10secretion.nat med.1999dec;5(12):1365-9.
14.9.chester c et al.immunotherapy targeting 4-1bb:mechanistic rationale,clinical results,and future strategies.blood.2018jan 4;131(1):49-57.
15.10.hamers-casterman c,et al.naturally occurring antibodies devoid of light chains.nature.1993jun 3;363(6428):446-8.
技术实现要素:
16.针对上述问题与不足,本技术提供一种工艺简单、成本低、特异靶向4-1bb的纳米抗体,可用于检测和制药领域。
17.本发明为了解决上述问题,提供的技术方案之一为:提供一种抗4-1bb纳米抗体,是如下a)或b)的蛋白:
18.a)由序列表中序列1所示的氨基酸序列组成的蛋白质;
19.b)在序列表中序列1的氨基酸序列经过取代和/或缺失和/或添加一个或几个氨基酸且与特异性识别4-1bb相关的由a)衍生的蛋白质。
20.优选的,所述由a)衍生的蛋白质为序列2-33所示的氨基酸序列组成的蛋白质。
21.具体的,本发明的抗4-1bb纳米抗体,其序列相似性>82.98%。根据氨基酸序列的相似性将33条序列分为6组,具体如下。
22.第一组包括24个序列,分别为:
23.p1-1a、p1-1f、p1-7d、p1-1c、p1-10b、p2-2e、p2-8a、p2-9h、p2-10h、p2-9c、p2-3d、p2-7f、p2-3f、p1-6h、p2-3c、p1-1g、p1-8b、p2-2h、p2-8g、p1-1b、p2-4b、p1-4f、p2-11d、p2-12g,分别对应序列表中序列1-24。其编码基因,分别对应序列表中序列34-57。
24.第二组包括2个序列,分别为:p2-7d、p1-4b,分别对应序列表中序列25和26。其编码基因,分别对应序列表中序列58和59。
25.第三组包括3个序列,分别为:p1-1e、p1-12h、p2-11h,分别对应序列表中序列27-29。其编码基因,分别对应序列表中序列60-62。
26.第四组包括1个序列,为p2-5a对应序列表中序列30。其编码基因,对应序列表中序列63。
27.第五组包括1个序列,为p2-10f对应序列表中序列31。其编码基因,对应序列表中序列64。
28.第六组包括2个序列,分别为:p1-2f、p2-1d,分别对应序列表中序列32和33。其编码基因,分别对应序列表中序列65和66。
29.将抗4-1bb纳米抗体进行一些修饰和改造,例如进行人源化、peg化或其他改造以提高其活性。
30.对此纳米抗体的cdr区域进行突变修饰,例如取代和/或缺失和/或添加一个或几个氨基酸,以提高其亲和力。
31.优选的,本发明的抗4-1bb纳米抗体还包括sp34-lc区域和sp34-hc区域,这两段区域共同组成cd3-fab,cd3-fab与抗4-1bb纳米抗体的融合构建,一方面促进纳米抗体的表达产量、另一方面提供tcr主要刺激信号,为后续t细胞活化验证提供帮助。本发明的抗4-1bb纳米抗体通过(g4s)3连接肽连接到sp34-lc的c端,形成以cd3-fab为骨架的融合抗体结构。该结构有利于验证本发明的抗4-1bb纳米抗体的独特优势,并且在验证中以cd3-fab为对照组说明了本发明的抗4-1bb纳米抗体的辅助功能特性。sp34-lc区域的氨基酸序列,对应序列表中序列67,其编码基因为对应序列表中序列70。(g4s)3连接肽区域的氨基酸序列,对应序列表中序列68,其编码基因为对应序列表中序列71。sp34-hc区域的氨基酸序列,对应序列表中序列69,其编码基因为对应序列表中序列72。
32.将抗4-1bb纳米抗体进行一些修饰和改造,例如进行人源化、peg化或其他改造以提高其活性。
33.对此纳米抗体的cdr区域进行突变修饰,例如取代和/或缺失和/或添加一个或几个氨基酸,以提高其亲和力。本发明为了解决上述问题,提供的技术方案之二为:提供该抗4-1bb纳米抗体的编码基因。
34.优选的,所述编码基因是如下1)或2)或3)的基因:
35.1)其核苷酸序列是序列表中序列34;
36.2)在严格条件下与序列34限定的dna片段杂交且编码与特异性识别4-1bb相关蛋白的dna分子;
37.3)与1)或2)的基因具有90%以上的同源性,且编码与特异性识别4-1bb相关蛋白的dna分子。
38.优选的,2)或3)中的基因,其核苷酸序列是序列表中序列35-66。
39.本发明为了解决上述问题,提供的技术方案之三为:提供含有该编码基因的重组表达载体或者重组菌株。
40.本发明为了解决上述问题,提供的技术方案之四为:提供该抗4-1bb纳米抗体,在制备预防或治疗肿瘤、免疫缺陷疾病以及感染性疾病等药物中的应用。
41.实施本发明,具有如下有益效果:本发明提供的抗4-1bb纳米抗体能够与4-1bb高效、特异性地结合,亲和力高达0.79nm,由于可以获得完整的抗体序列,纳米抗体能够通过体外重组表达从而高质量、稳定的生产,具有广泛的应用前景。
42.本发明的上述应用中,包括如下中的至少一种:
43.(1)直接应用于4-1bb抗原特异性结合,例如以4-1bb为靶点的肿瘤抗原特异性识别与杀伤;
44.(2)在本发明所述4-1bb纳米抗体上连接效应分子,例如化学小分子药物、细胞因子、毒性分子或放射性同位素,用于医药领域;
45.(3)以所述4-1bb纳米抗体作为药物递送载体,定点靶向肿瘤等抗原部位,以实现定点特异性治疗;
46.(4)在本发明所述4-1bb纳米抗体上添加化学发光基团或荧光分子,以制备特异性探针;
47.(5)将发明所述4-1bb纳米抗体用于免疫系统增强疗效,例如car-t,tcr-t,car-nk,car-dc等,用于免疫治疗领域。
附图说明
48.图1 4-1bb抗原蛋白sds-page鉴定结果;
49.图2羊驼抗4-1bb血清效价检测结果;
50.图3羊驼pbmc总rna琼脂糖凝胶电泳结果;
51.图4阳性菌落pcr琼脂糖凝胶电泳结果;
52.图5一轮阳性菌落酶联免疫吸附实验结果,包括图5a~5q;
53.图6二轮阳性菌落酶联免疫吸附实验结果,包括图6a~6d;
54.图7抗4-1bb纳米抗体序列相似性示意图,包括图7a~7e,图7a显示出了所列实施例中序列的相似性,图7b~7e显示出了组间序列的相似性;
55.图8cd3/4-1bb融合抗体的elisa检测结果,包括图8a和8b,其中图8a为cd3/4-1bb融合抗体和对照cd3抗体与靶抗原4-1bb抗原结合能力检测结果,图8b为cd3/4-1bb融合抗体和对照cd3抗体与不相关抗原cd28结合能力检测结果;
56.图9 cd3/4-1bb融合抗体与k562细胞表面受体的结合实验结果,包括图9a和9b,其中图9a为cd3/4-1bb融合抗体和对照cd3抗体与靶细胞k562/4-1bb的结合能力检测结果,图9b为cd3/4-1bb融合抗体和对照cd3抗体与不相关细胞k562/cd28的结合能力检测结果;
57.图10 cd3/4-1bb融合抗体对人pbmc的活化结果。
具体实施方式
58.下面结合附图和具体实施例对本发明的技术方案进行详细的说明,但是所述实施例的说明,仅仅是本发明的一部分实施例,其中大部分并不仅限于此。
59.由于单克隆抗体的筛选及获得技术难度大且成本高,因此我们将4-1bb免疫羊驼并获得其纳米抗体文库,通过噬菌体展示库技术,筛选获得高亲和力纳米抗体。本抗体可应用于肿瘤、免疫缺陷疾病和感染性疾病的免疫治疗,并通过酶联吸附反应等对4-1bb蛋白进行定性和定量测定。
60.尽管4-1bb蛋白是肿瘤免疫疗法的潜在靶点,现有的单克隆抗体在临床试验中呈现出了肝毒性、疲劳、发热等不良反应,而安全性较高的utomilumab对受体的激动活性则较低。因此开发新的4-1bb靶向抗体药物具有积极意义,并为后续单独用药或药物联用提供新的思路。相比于常规抗体,本发明所述4-1bb纳米抗体的优势有:
61.(1)分子量小,可穿透血脑屏障,增强病灶特异性渗透,从而提升靶向杀伤抗原的效果;
62.(2)在原核或真核系统中表达高;
63.(3)稳定性好,特异性强,亲和力高;
64.(4)不会对人体引起免疫原性反应。
65.本发明所述抗4-1bb纳米抗体的应用优势在于生物医药研发(基因工程药物研发、adc药物研发);临床体外诊断(胶体金法、酶联免疫吸附法、电化学发光法);肿瘤研究、免疫学治疗研究等基础研究,以及探针等特异性靶向识别与杀伤等方面的应用。
66.实施例1 4-1bb蛋白的表达及纯化
67.1.1载体构建
68.以真核表达载体pfuse构建4-1bb-fc融合蛋白,将4-1bb和人igg1 fc通过pcr方式串联起来,完整分子包括4-1bb分泌信号肽、4-1bb胞外结构域、凝血酶切位点、iegrmd短肽、人igg1铰链区以及ch2和ch3结构域。将4-1bb-fc基因采用ncoi和nhei双酶切后使用t4 dna连接酶克隆至pfuse载体,或者采用同源重组试剂盒构建载体。
69.1.2真核表达
70.接种1.5
×
106/ml 293f细胞于500ml摇瓶200ml培养基中。37℃,165rpm,5%co2浓度的摇床培养箱中振摇培养24h,24h后进行细胞计数,将细胞密度调整至3
×
106/ml,将上述1.1步骤所构建的质粒加200μg于10ml opti-mem轻柔混匀后室温静置5min,加入500μl(1:2.5)pei 40000于10ml opti-mem轻柔混匀后室温静置5min,混合质粒和pei混合液,轻柔混匀后室温静置20min。将上述混合液逐滴加入200ml 293f细胞培养液中,滴加的同时轻柔摇晃培养瓶,混合均匀后放入摇床,转染72h。转染结束后,100g离心5min,取上清液,向培养瓶中补加100ml培养基重新悬浮培养后3000rpm离心5min,取上清液。混合两次细胞上清液。
71.1.3蛋白纯化
72.取上述1.2步骤的细胞上清液400ml,15000rpm,4℃离心30min,收取上清液,0.45μm滤膜过滤,置于冰上备用。取3ml(20%乙醇/protein g 1:1)protein g于层析柱内,用binding buffer冲洗3次后,使用垫片压住resin表面。用20ml binding buffer平衡protein g柱。每10ml上样一次,使样品匀速(约0.5ml/min)通过protein g柱。40ml binding buffer匀速(约1ml/min)清洗protein g柱。首先在洗脱管收集管中加入10%洗脱液体积的buffer,柱中加入elution buffer(5ml洗脱一次,直到无法定量蛋白浓度)洗脱4次。使用amicon ultra-15离心过滤器对收集的蛋白样品进行浓缩,4℃离心机3000rpm离心20分钟,并使用nanodrop测蛋白浓度。
73.将蛋白样品进行缓冲液置换。放入透析袋(加超纯水微波炉煮十分钟)中,4℃,4l透析液(转子搅拌透析),每4小时换一次液。两次换液后,收集透析袋内全部蛋白液体,4℃,8000rpm,15分钟离心,收集上清并定量。
74.首先进行酶切效率测试,取适量定量后的蛋白溶液,浓度稀释为1mg/ml,每管取100μl蛋白样品。加thrombin(凝血酶),浓度从10iu/mg开始,2倍稀释(使用透析液),共10个梯度。设置不加thrombin的对照组。室温振荡16小时。sds-page检测酶切效果。确定合适thrombin酶切浓度,将剩余蛋白样品全部酶切。室温振荡16小时。sds-page检测大体系酶切效果。
75.将凝血酶酶切后蛋白样品缓慢匀速通过protein g层析柱,并按照上述洗脱方法
进行洗脱,并用sds-page进行检测,直至样品中没有fc,4-1bb ecd纯度大于90%为佳。
76.浓缩除标签后的4-1bb ecd蛋白至1mg/ml,并取样进行sds-page进行最终鉴定(4-1bb ecd-fc分子量约为44.8kd,由于真核表达的糖基化作用,还原型sds-page最终表现应为50-65kd,fc分子量约为30kd),结果如图1所示。
77.实施例2抗4-1bb纳米抗体噬菌体展示文库的构建及筛选
78.2.1羊驼的免疫
79.选取健康成年羊驼一只,标记耳号。将4-1bb蛋白与弗氏佐剂按1:1的比例混合均匀,4℃保存。在羊驼颈部淋巴结附近分左右两侧注射,每侧分2点,每次注射0.4ml混合佐剂抗原。免疫后观察半小时,确认羊驼状态良好且无不良反应。每2周免疫一次,一共进行7次免疫。第6、7次免疫后间隔5-7天,颈部静脉采集羊驼外周血,用于噬菌体展示文库构建。
80.2.2羊驼来源的淋巴细胞分离
81.在15ml离心管中加入3ml细胞分离液,缓慢加入3ml血样。离心机25℃预冷,400g离心30min,观察血样分离情况。使用200μl移液器小心吸取中间棉状上层免疫细胞至新的15ml离心管中,上层血清转移至新离心管后-80℃保存。向含免疫细胞的离心管中加入10ml室温pbs缓冲液,25℃,400g离心20min,去除上清液。每管加入5ml室温pbs缓冲液,25℃,400g离心20min,计数,去上清液。根据细胞计数结果使用rnaiso plus溶解分离得到的淋巴细胞,得到溶解液,-80℃保存。
82.2.3羊驼血清效价检测
83.抗原包被酶标板(2μg/ml,100μl/孔),4℃过夜。弃抗原溶液,并用200μl pbst溶液(1
×
pbs加0.05%tween20,下同)冲洗酶标板三次,每次5分钟。使用200μl封闭液(2%bsa in pbst)封闭酶标板,室温,1.5小时。弃封闭液,并用200μl pbst溶液冲洗酶标板三次,每次5分钟。使用封闭液对各批次血清进行梯度稀释,并向酶标板对应孔中加入100μl相应的血清,室温孵育2小时。弃血清,并用200μl pbst溶液冲洗酶标板4次,每次5分钟。向每个孔中加入检测抗体(山羊抗美洲驼igg,偶联hrp,1:10000稀释于封闭液中,100μl/孔)室温,1小时。弃检测抗体,并用200μl pbst溶液冲洗酶标板4次,每次5分钟。向每孔加入100μl tmb底物溶液,显色2-3分钟。向每孔加入100μl反应终止液。30分钟内使用酶标仪测定450nm处吸光值。阳性孔标准:免疫血清样品od
450
值大于未免疫血清3倍且读值大于0.5,结果如图2所示。
84.2.4rna提取
85.将trizol保存的外周血淋巴细胞转移至1.5ml离心管中,加入1/5体积的氯仿混匀,室温静置5min,4℃,12000g离心15min。小心移取上清液至新的离心管中,加入0.5-1倍体积的异丙醇,室温静置10min,4℃,12000g离心10min。使用trizol保存的外周血淋巴细胞等体积的75%乙醇清洗沉淀,4℃,7500g离心5min后,溶解于适量rnase-free的水中,合并所有样品,即为提取得到的总rna。羊驼pbmc总rna琼脂糖凝胶电泳结果如图3所示。
86.2.5反转录合成cdna
87.使用takara反转录试剂盒对获得的总rna进行反转录。将上述总rna样品分为两份,一份使用试剂盒内的oligo dt primer作为引物,另一份用试剂盒内的random 6-mers作为引物,按照反转录试剂盒说明书将上一步得到的总rna反转录成cdna,分别保存到2个离心管中。
88.2.6抗体可变区基因扩增
89.将反转录得到的cdna作为模板,使用taq dna polymerase hot start酶进行两轮pcr扩增反应。
90.第一轮反应条件和程序为:
91.使用taq dna polymerase hot start酶进行pcr扩增,为了确定最佳模板使用量,分别使用1μl、2μl、3μl、4μl、5μl的oligo dt primer和random 6-mer的cdna作为模板,按照下表配置pcr反应体系:
[0092][0093]
按照下表配置并进行pcr反应:
[0094][0095]
(1)反应结束后,取20μlpcr产物进行1%琼脂糖凝胶电泳,最终选取电泳结果中目的条带单一且片段大小为600bp的模板量为最佳模板量,将所有cdna按照此模板量并使用相同条件进行pcr反应;
[0096]
(2)将所有pcr产物进行1%琼脂糖凝胶电泳,并切胶回收目的片段大小在600bp左右的条带;
[0097]
(3)将所有一轮pcr纯化回收产物收集至一个离心管中,即为第一轮pcr扩增产物,-20℃保存。
[0098]
第二轮反应条件和程序为:
[0099]
将第一轮pcr扩增产物作为模板进行第二轮pcr反应,为了确定最佳模板使用量,分别使用0.5μl、1μl、2μl、3μl、4μl作为模板,按照下表配置pcr反应体系:
[0100]
[0101][0102]
按照下表配置并进行pcr反应:
[0103][0104]
(1)反应结束后,取20μl pcr产物进行1%琼脂糖凝胶电泳,最终选取电泳结果中目的条带单一且片段大小为600bp的模板量为最佳模板量,将已获得的第一轮pcr扩增产物的约1/5体积共288个反应按照此模板量并使用2.5第一轮扩增相同条件进行pcr反应后使用通用型dna纯化回收试剂盒对pcr反应液进行dna纯化;
[0105]
(2)将回收产物收集至一个离心管中,即为第二轮pcr扩增产物,同时取2μl用核酸浓度测量仪检测回收产物的浓度并记录,其余产物-20℃保存。
[0106]
2.7载体构建
[0107]
(1)使用padl-10b作为噬菌体质粒载体,用bgli分别酶切10μg padl-10b载体和5μg第二轮pcr扩增产物,37℃孵育4h;
[0108]
(2)使用dna回收纯化试剂盒对padl-10b载体和第二轮pcr扩增产物进行纯化,4℃保存。
[0109]
2.8连接
[0110]
(1)载体和片段进行连接,按照下表配制连接反应体系:
[0111][0112]
(2)将连接反应4℃过夜(约16h)孵育;
[0113]
(3)使用通用型dna纯化回收试剂盒纯化连接反应液,检测回收产物的浓度,4℃保存。
[0114]
2.9验证连接产物转化率
[0115]
(1)取一支50μl的ss320感受态细胞在冰上放置5-10min融化;
[0116]
(2)加入100ng连接产物,转移到已经预冷好的间距1mm的电转杯中,在电转仪中设定参数:1800v,1mm后,点击按钮点击转化;
[0117]
(3)等待电转完成后立即加入37℃预热好的soc培养液1ml,混匀后37℃,200rpm摇
菌复苏1h;
[0118]
(4)从1ml复苏后的菌液中取100μl进行10倍梯度稀释并涂板,根据稀释倍数和单菌落数计算每个反应能获得的转化菌落数目,即为连接产物的转化效率;
[0119]
(5)同时随机挑选48个单克隆菌进行菌落pcr,pcr产物有在500bp左右的单一条带认为是阳性克隆,由此估算单克隆阳性率,结果如图4所示。
[0120]
2.10细菌文库构建
[0121]
(1)取25组100ng连接体系按上述方法使用25管感受态细胞进行电转反应;
[0122]
(2)37℃复苏1h后从中取100μl进行10梯度稀释后涂板,37℃过夜培养;
[0123]
(3)收集剩下所有菌液并均匀涂布到5个245mm方形培养板(2
×
yt含有100μg/ml amp,2%glucose,2%agarose)中,37℃培养过夜;
[0124]
(4)根据稀释倍数和单菌落数计算所有反应能获得的转化菌落数目,即为细菌文库的库容量;
[0125]
(5)同时从梯度稀释板中随机挑选48个单克隆进行菌落pcr,验证细菌文库的克隆阳性率;
[0126]
(6)将过夜培养的245mm方形培养板菌落使用2
×
yt液体培养基刮下,置于50ml离心管,测量其od
600
值,添加终浓度20%甘油-80℃保存。
[0127]
2.11噬箘体文库制备
[0128]
根据细菌文库的od
600
计算在100ml 2
×
yt液体培养基需要加入的细菌文库体积,据计算结果接种细菌文库到100ml 2
×
yt液体培养基(含有10μg/μl tet和100μg/μl amp)中,37℃,250rpm培养直至od
600
为0.5-0.55。根据辅助噬菌体效价,按照1:20(细菌个数:噬箘体数)的比例加入辅助噬菌体,37℃,250rpm孵育30-60min。加入终浓度为50μg/ml和200μm的iptg,30℃,250rpm过夜培养。将过夜培养的菌液4℃,8000rpm离心10min,然后将离心后的上清液转移到新的50ml离心管中。加入1/4的预冷peg/nacl储存液,混合均匀,冰上静置孵育30min。4℃,8000rpm离心10min后,弃上清,倒置控干2min。加入5ml pbs重悬至新的离心管,4℃,8000rpm离心10min。离心后转移上清至新的离心管中,再次加入1/4体积的预冷peg/nacl储存液,混合均匀,冰上孵育10min。4℃,8000rpm,10min离心,弃上清,用1ml pbs重悬后,8000rpm离心10min,转移上清至新的离心管,-80℃保存,即为纯化后的噬菌体文库。
[0129]
2.12阳性噬菌体库筛选
[0130]
第一轮筛选:从-80℃冰箱中取出筛选抗原,冰上放置解冻;将筛选抗原4-1bb及负筛抗原cd28分别包被免疫管(50μg/管,包被液为pbs,2ml/管),其中负筛抗原包被两个免疫管,一管做负筛,一管做对照,4℃缓慢旋转包被过夜。将过夜包被的免疫管中的液体弃掉,加入2ml pbs缓冲液室温清洗免疫管3次,每次旋转5min。加入2ml封闭液(3%脱脂奶粉)溶液,室温旋转封闭2h。先将封闭后的负筛免疫管中液体丢弃,并加入2ml pbst缓冲液室温清洗免疫管3次,每次旋转5min。弃掉负筛免疫管中的清洗液,加入2ml pbs,按照下列公式计算并加入制备的噬菌体文库104μl作为第一轮筛选输入噬菌体文库,室温旋转孵育1h:
[0131]
[0132]
其中,v为加入的噬菌体体积(单位μl),t
library
为噬菌体效价;
[0133]
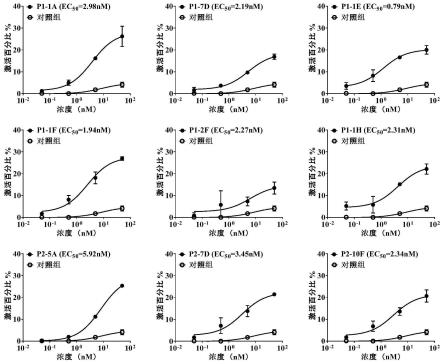
将包被抗原4-1bb和对照抗原cd28的免疫管内封闭液弃掉,加入2ml pbst(1
×
pbs加0.1%tween20,下同)缓冲液室温清洗免疫管3次,每次旋转5min。将免疫管内液体弃掉,将负筛孵育结束后的噬菌体溶液均分成分别加入这两个免疫管中,室温旋转孵育1h;弃掉免疫管中的液体,加入2ml pbst缓冲液室温清洗免疫管20次,每次旋转5min。将免疫管内液体弃掉,尽量去除残余液体,加入1ml 0.25mg/ml trypsin溶液,室温旋转洗脱30min。加入10μl 10%aebsf终止洗脱,将免疫管中的溶液转移至新的1.5ml离心管中,即为第一轮筛选噬菌体洗脱液。
[0134]
第一轮噬菌体洗脱液效价检测:将-80℃冰箱保存的ss320菌株在2
×
yt固体培养基(tet抗性)进行单菌落划线,37℃过夜培养(4℃保存一周),从单菌落板上挑取一个单菌落到5ml含有10μg/ml tet的2
×
yt培养基,37℃过夜培养。取过夜培养菌液500μl转接到含5ml含有10μg/ml tet的2
×
yt液体培养基中,37℃,250rpm培养约45min-60min后至od
600
为0.5-0.55。取第一轮噬菌体洗脱液10μl在1.5ml离心管中10倍梯度稀释,共稀释10个梯度,即将第一轮噬菌体洗脱液取10μl稀释至100μl,再从中取10μl稀释至100μl,依次类推,共稀释12个梯度至10-10
,振荡混匀。在每个稀释离心管中加入90μl的ss320菌液,混匀后37℃孵育30min,从每个稀释离心管中取5μl滴加到2
×
yt固体培养基(amp)中,37℃倒置过夜培养。统计可以明显区分单菌落的稀释度的平板上的单菌落数量,并按照下面公式计算每毫升菌体溶液中噬菌粒的数量,即噬菌体文库效价:
[0135]
t(pfu/ml)=n
×d×
400
[0136]
其中,t为噬菌体效价(单位pfu/ml),d为稀释倍数,n为相应稀释倍数上单菌落个数。
[0137]
第一轮噬菌体洗脱液的扩增:预先将-80℃冰箱保存的ss320菌株在2
×
yt固体培养基(tet抗性)进行单菌落划线,37℃过夜培养(4℃保存一周),从单菌落板上挑取一个单菌落到5ml含有10μg/ml tet的2
×
yt培养基,37℃过夜培养。取过夜培养菌液500μl转接到含5ml含有10μg/ml tet的2
×
yt液体培养基中,37℃,250rpm培养约45min-60min后至od
600
值为0.5-0.55。加入500μl第一轮筛选后得到的噬菌体洗脱液至od
600
为0.5-0.55的菌液中(剩余的洗脱液4℃保存)。37℃,250rpm继续培养30min。将全部菌液均匀涂布到含有100μg/mlamp和2%葡萄糖的2%琼脂糖的245mm方形培养基平板中,37℃过夜培养。取过夜培养的方形板,在培养板表面加入6ml 2
×
yt液体培养基(含10μg/ml tet),用涂布棒轻轻将方形板菌落刮下并将菌液收集至15ml离心管中,即为扩增后的细菌子文库,同时使用分光光度计测量菌液od
600
值,即为洗脱液细菌文库od
600
值,加入终浓度为20%的甘油,即为第一轮细菌文库。将洗脱液细菌文库根据下面公式计算出相应菌液量,转接至100ml 2
×
yt液体培养基中(含10μg/ml tet和100μg/mlamp),使初始od
600
为0.1:
[0138]
其中,v为转接菌液的体积(单位μl),od
600
为构建的洗脱液细菌文库的od
600
。
[0139]
37℃,250rpm培养直至菌液od
600
达到0.5-0.55。按照下面公式计算并加入辅助噬菌体m13k07以使细菌个数:噬菌体数=1∶20:
[0140][0141]
其中,v为加入辅助噬菌体的体积(单位ml),t
helper-phage
为使用的辅助噬菌体效价,od
600
为菌液的od
600
值;
[0142]
37℃,250rpm继续培养30min;分别加入终浓度为50μg/ml kana和终浓度为0.2μm iptg,30℃,250rpm过夜培养。
[0143]
第一轮噬菌体纯化:将过夜培养的菌液转移至新的50ml离心管中4000rpm,4℃离心10min。将离心后的上清液转移至新的50ml离心管中,加入1/4体积的4℃预冷的20%peg/2.5m nacl,充分混匀后冰上放置30min。4000rpm,4℃离心20min,弃上清,并在纸上倒置2min。加入1mlpbs重悬沉淀,将重悬液移至新的1.5ml离心管,13000rpm,4℃离心20min。将离心后的上清转移至新的1.5ml离心管,加入1/4体积预冷的20%peg/2.5mnacl溶液,混匀后冰上放置10min。13000rpm,4℃离心10min,弃上清,加入1ml pbs重悬沉淀,13000rpm,4℃离心2min,将上清转移到新的1.5ml离心管中,即为第一轮筛选噬菌体子文库,100μl/管分装,长期-80℃保存,短期(1-2周)可于-20℃放置保存。一轮筛选噬菌体子文库效价检测,方法同上。
[0144]
第二轮筛选:筛选方法同上,输入噬菌体为第一轮筛选得到噬菌体子文库,作为第二轮筛选输入噬菌体文库,得到第二轮筛选噬菌体洗脱液。
[0145]
第二轮噬菌体洗脱液效价检测、洗脱液的扩增、纯化、筛选噬菌体子文库效价检测,方法同上。
[0146]
单克隆elisa检测:预先将-80℃冰箱保存的ss320菌株在2
×
yt固体培养基(tet抗性)进行单菌落划线,37℃过夜培养(4℃保存一周),从单菌落板上挑取一个单菌落到5ml含有10μg/mltet的2
×
yt培养基,37℃过夜培养。取过夜培养菌液500μl转接到5ml含有10μg/mltet的2
×
yt液体培养基中,37℃,250rpm培养45min-60min后至od
600
值为0.5-0.55。取第二轮筛选后噬菌体洗脱液10μl,在1.5ml离心管中10倍梯度稀释,共稀释12个梯度,振荡混匀。每个稀释离心管中加入90μl od
600
值为0.5-0.55的菌液,混合均匀。37℃,250rpm继续培养30min。将菌液均匀涂布到含有100μg/mlamp的固体培养基平板,37℃过夜培养。从过夜培养后的培养基平板中随机挑取单克隆菌落于无菌96孔细胞培养板(p1-p2)中,每孔加入100μl 2
×
yt培养基(含有100μg/ml amp和10μg/ml tet),37℃过夜静置培养。取过夜培养的菌液2μl转接至每孔250μl 2
×
yt液体培养基(含有100μg/mlamp和10μg/ml tet)的新96孔细胞培养板中,37℃静置培养3h,将转接前过夜培养的菌液4℃保存。按照下面公式计算并每孔加入辅助噬菌体m13k07以使细菌个数:噬菌体数=1∶20:
[0147][0148]
其中,v为加入辅助噬菌体的体积(单位ml),t
helper-phaqe
为使用的辅助噬菌体效价。
[0149]
37℃孵育30min,加入终浓度为50μg/ml的kana和0.2μm的iptg,30℃静置过夜培养。将过夜培养后的96孔培养板4℃,4000rpm离心10min,4℃保存备用。将筛选抗原4-1bb及负筛抗原cd28分别包被酶标板(1ng/μl,包被液为ph 9.4的cbs,100μl/孔),同时平行包被bsa作对照,4℃包被过夜。将过夜包被的酶标板内液体弃掉,每孔加入200μl pbs缓冲液,室
温清洗酶标板3次,每次10min。每孔加入200μl封闭液(3%bsa in pbst)封闭酶标板,室温封闭1h。弃封闭液,每孔加入200μl pbst(1
×
pbs加0.1%tween20,下同)缓冲液,室温清洗酶标板3次,每次10min。每孔加入120μl 3%bsa后再加入80μl离心后的上清液,室温孵育2h。弃掉酶标板内液体,每孔加入200μl pbst缓冲液洗涤3次,每次10min。每孔加入m13 bacteriophageantibody(hrp),mouse mab,1:8000稀释于封闭液中,100μl/孔,室温孵育1h。弃掉elisa板内液体,每孔加入200μl pbst缓冲液洗涤3次,每次10min。每孔加入100μl tmb单组份显色液避光显色2-3min,每孔加入100μl1m hcl终止,用酶标仪读取od
450
值,记录并保存。一次验证以负筛抗原4-1bb、bsa做阴性对照,以od
600
≥0.6136为阳性克隆范围。结果使用graphpad prism处理,如图5a~5i所示。
[0150]
阳性克隆elisa二次验证:为了排除假阳性结果,将初步认定为阳性的克隆进行elisa二次验证,方法同上,二次验证只做负筛抗原cd28对照,不做bsa对照。测序结果具有代表性的阳性单克隆二次验证,以od
600
≥0.6037为阳性克隆范围。结果使用graphpad prism处理,结果如图6a~6b所示。
[0151]
实施例3测序
[0152]
3.1阳性克隆测序
[0153]
根据elisa检测数据和二次验证数据选定阳性单克隆。
[0154]
从单克隆elisa检测板中取阳性克隆菌液5μl接种至2ml 2
×
yt培养基(含有100μg/ml amp和10μg/ml tet),37℃,250rpm培养直至od
600
至0.8-1.0(约6-8h),取1ml菌液测序,其余菌液4℃保存。
[0155]
3.2序列分析
[0156]
经测序的序列使用gentle软件进行序列比对分析,并使用gentle软件将抗体序列翻译成氨基酸。结果如图7a~7e,其中图7a显示出所列实施例中总序列相似性,图7b~7e显示出组间序列相似性。
[0157]
实施例4 cd3/4-1bb融合抗体的表达和纯化
[0158]
4.1载体构建
[0159]
以真核表达载体pfuse构建sp34-hc和sp34-lc-4-1bb(nanobody)融合蛋白。对于sp34-hc,完整分子包括il2分泌信号肽和sp-34-vh和sp-34-ch1;对于sp34-lc-4-1bb(nanobody)的构建,将sp34-lc和抗4-1bb纳米抗体通过pcr方式串联起来,完整分子包括il2分泌信号肽、sp34-lc、(g4s)3连接肽、抗4-1bb纳米抗体。将sp34-hc或者不同的sp34-lc-4-1bb(nanobody)基因采用ecori和xhoi双酶切后使用t4 dna连接酶克隆至pfuse载体,或者采用同源重组试剂盒构建载体。
[0160]
4.2真核表达
[0161]
接种1.5
×
106/ml 293f细胞于250ml摇瓶50ml培养基中。37℃,165rpm,5%co2浓度的摇床培养箱中振摇培养24h,24h后进行细胞计数,将细胞密度调整至3
×
106/ml,将上述4.1步骤所构建的质粒sp34-hc和sp34-lc-4-1bb(nanobody)(25μg:25μg)于5ml opti-mem轻柔混匀后室温静置5min,加入125μl(1:2.5)pei 40000于10ml opti-mem轻柔混匀后室温静置5min,混合质粒和pei混合液,轻柔混匀后室温静置20min。将上述混合液逐滴加入50ml 293f细胞培养液中,滴加的同时轻柔摇晃培养瓶,混合均匀后放入摇床,转染72h。转染结束后,100g离心5min,取上清液,向培养瓶中补加50ml培养基重新悬浮培养后3000rpm
离心5min,取上清液。混合两次细胞上清液。
[0162]
4.3蛋白纯化
[0163]
取上述1.2步骤的细胞上清液50ml,15000rpm,4℃离心30min,收取上清液,0.45μm滤膜过滤,置于冰上备用。取3ml(20%乙醇/protein g 1:1)protein g于层析柱内,用binding buffer冲洗3次后,使用垫片压住resin表面。用20ml binding buffer平衡protein g柱。每10ml上样一次,使样品匀速(约0.5ml/min)通过protein g柱。40ml binding buffer匀速(约1ml/min)清洗protein g柱。首先在洗脱管收集管中加入10%洗脱液体积的buffer,柱中加入elution buffer(5ml洗脱一次,直到无法定量蛋白浓度)洗脱4次。使用amicon ultra-15离心过滤器对收集的蛋白样品进行浓缩,4℃离心机3000rpm离心20分钟,并使用nanodrop测蛋白浓度。
[0164]
实施例5 cd3/4-1bb融合抗体与4-1bb抗原的结合能力验证
[0165]
5.1实验开始前,各试剂均应平衡至室温;试剂或样品配制时,均需充分混匀,并尽量避免起泡。
[0166]
5.2包被:用包被液4℃过夜包被4-1bb抗原(10ug/ml),分为四组,每组两个平行,一个空白对照。同时包被一个阴性对照组,以抗原cd28(10ug/ml)包被阴性对照组。
[0167]
5.3封闭:每孔加封闭液200ul,37℃封闭1h。
[0168]
5.4加样:分别设空白孔、待测样品孔。所有样品均用封闭液稀释。首先配置150ul(两个平行)的200nm的4-1bb抗体(使用封闭液稀释抗体),从每个平行组200nm抗体中取30ul,加入到120ul的下一孔稀释液中(5倍稀释)。以200nm为起始浓度5倍梯度稀释,7孔(100ul/孔),空白对照孔只加封闭液。盖上酶标板,37℃孵育90分钟(室温2h)。
[0169]
5.5孵育二抗:先弃去液体(在纸上用力拍干),用200ul/孔pbst洗涤重复3次,每次放水平摇床3-5min。每孔加入hrp-anti kappa light chain antibody二抗100μl,加上覆膜,37℃温育1h。
[0170]
5.6显色:弃去孔内液体,甩干,洗板5次,每孔加显色底物tmb 100μl,酶标板加上覆膜37℃避光孵育。每孔加终止液2m h2so
4 50μl,终止反应,此时蓝色立转黄色。立即用酶标仪在450nm波长测量各孔od值。应提前打开酶标仪电源,预热仪器,设置好检测程序。
[0171]
5.7依据od值绘制标准曲线,结果如图8a和8b所示。实验组有明显的抗原结合能力,与对照组有显著差异。
[0172]
实施例6 cd3/4-1bb融合抗体与k562/4-1bb细胞的结合实验
[0173]
6.1实验组做两个平行100μl/孔,阴性对照组做两个平行。按蛋白浓度梯度把每组分为7个点。阳性细胞k652/4-1bb cells:(0.3million/孔),每组7孔;阴性细胞k562/cd28cells:(0.3million/孔),每组7孔。
[0174]
6.2蛋白浓度梯度稀释:在96孔板中,将4-1bb蛋白稀释到200nm的初始浓度,五倍梯度稀释分别为(200nm、40nm、8nm、1.6nm、320pm、64pm、12.8pm)。等待处理好细胞后,向细胞中加入100ul已经做好浓度梯度稀释的抗体。
[0175]
6.3将k562细胞计数,调整每孔细胞量为3
×
105将细胞放入u型孔板,离心去上清,加入200μl facs重悬细胞,离心去上清。
[0176]
6.4将步骤2稀释的蛋白按照100μl/组,加入不同的k562细胞中,放冰上孵育2h后,加入facs清洗掉多余的游离抗体,离心去掉上清。
[0177]
6.5加入稀释好的goat anti-human kappa-af467 100μl,吹打重悬细胞置于冰上孵育1h.之后离心去掉上清。加入200μl facs,清洗掉多余的游离抗体,离心去掉上清,再用200μlfacs缓冲液重悬细胞。
[0178]
6.6流式检测:流式检测数据使用graphpad prism处理,结果如图9a和9b所示。实验组有明显的细胞表面抗原结合能力,与对照组有显著差异。
[0179]
实施例7 cd3/4-1bb融合抗体对人pbmc的活化能力验证
[0180]
7.1本轮实验使用pbmc进行活化,实验均为两组平行,每组均设置空白对照,cd3-fab作为阴性对照。
[0181]
7.2细胞计数,调整pbmc细胞密度为1
×
106/ml,48孔板每孔2.5ml;
[0182]
7.3 cd3/4-1bb融合抗体:配置激活剂浓度梯度,三个梯度,两个平行,每孔加入50nm、5nm、0.5nm和0nm cd3/4-1bb抗体,100μl/孔。
[0183]
7.4按浓度每孔加入细胞体积1/10抗体(20μl),加入cd3/4-1bb抗体。
[0184]
7.5 37℃培养48h后离心1000rpm离心5min,进行流式cd25/cd69染色,结果如图10所示。实验组有明显的t细胞活化能力,与对照组有显著差异。
[0185]
以上文字及附图对本发明进行了描述,但是本发明并不局限于上述的具体实施方式,上述具体实施方式仅起到示意作用,不具有限制性。
[0186]
本发明根据序列相似性比对结果,以组间相似性>93%将筛选出的33条序列分为6组,每组选出代表性序列进行功能验证:(1)4-1bb抗原结合能力检测;(2)细胞表面抗原结合能力检测;(3)t细胞活化能力检测。本发明设计的抗4-1bb纳米抗体以cd3-fab为骨架,因此抗体功能验证实验以cd3-fab作为阴性对照。本发明设计的抗4-1bb纳米抗体有较好的4-1bb抗原结合能力、细胞表面抗原结合能力以及对人pbmc细胞的活化能力,并且与cd3-fab阴性对照组有明显差异。
[0187]
以上实施例进一步说明本发明的内容,但不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改和替换,均属于本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。