1.本发明是涉及键合有在细胞内进行分解的分支型分解性聚乙二醇衍生物的生物相关物质的发明。
背景技术:
2.使用激素、细胞因子、抗体、酶等生物相关物质的药品,通常在给药至生物体内时,由于肾脏中的肾小球过滤、肝脏或脾脏等中的巨噬细胞的摄取,迅速从生物体内被排出。因此,多存在血液半衰期缩短、难以获得充分的药理效果的情况。为解决该问题,已经进行了通过糖链、聚乙二醇等亲水性高分子或白蛋白等对生物相关物质进行化学修饰的尝试。其结果是由于分子量的增大、水合层的形成等,能够延长生物相关物质的血液半衰期。此外,通过以聚乙二醇进行修饰,可获得生物相关物质的毒性或抗原性的降低、难水溶性的药剂的溶解性提高等效果也广为人知。
3.被聚乙二醇修饰的生物相关物质由于被由聚乙二醇的醚键与水分子的氢键所形成的水合层覆盖,分子尺寸增大,因此可避免肾脏中的肾小球过滤。进一步地已知其与调理素、构成各组织的细胞表面之间的相互作用降低,向各组织中的迁移减少。已知聚乙二醇是能够延长生物相关物质的血液半衰期的优异材料,其性能是分子量越大则效果越高。迄今为止,较多地进行了以分子量4万以上的高分子量的聚乙二醇进行修饰的生物相关物质的研究,获得了能够显著地延长其血液半衰期的结果。
4.聚乙二醇被作为用于生物相关物质的性能改善的修饰剂中的最佳标准,现在已经有多个聚乙二醇修饰制剂上市,并用于医疗场所。另一方面,2012年欧洲药品管理局(ema)报道了,在长时间用规定的给药量以上对动物给药以分子量4万以上的高分子量的聚乙二醇修饰的生物相关物质时,一部分的组织的细胞内将产生空泡这样的现象(非专利文献1)。当前,考虑到尚无空泡的产生本身对人体带来不良影响的报道,此外,上述ema的报道中所使用的给药量,相比于医疗场所中通常所适用的给药量为极高的用量等,可以说目前制造销售的分子量4万以上的聚乙二醇所修饰的治疗制剂的安全性没有问题。然而,也能够设想到在非常特殊的疾病(例如侏儒症等)的治疗中,采用高用量且长时间向患者给药聚乙二醇修饰制剂的治疗方案。因此,预计在开发这样的特殊状况中也能够适用的、在细胞中不产生空泡的聚乙二醇修饰制剂方面存在潜在需求。
5.非专利文献2中,在将相比于通常的聚乙二醇修饰制剂的给药量大幅过量的聚乙二醇,长时间单独向动物给药时,在分子量2万时未发现空泡,而在分子量4万时确认到空泡的产生。作为抑制空泡的一个手段,想到缩小聚乙二醇的分子量,但产生如果缩小分子量则不能充分改善生物相关物质的血液半衰期这一问题。
6.关于促进高分子量的聚乙二醇在体内分解为低分子量的聚乙二醇,从肾脏排出的技术,有报道例子。专利文献1中记载了具有能在生物体内被切断的硫醚键或肽键部位的聚乙二醇衍生物。记载了该聚乙二醇衍生物在生物体内被分解至适于从肾脏排出的分子量。然而,完全没有显示出有关具体分解的数据,也没有自肾脏的排出得到促进这样的数据。进
一步地,没有关于细胞的空泡的记载。
7.专利文献2中记载了具有在生物体内的低ph环境下能够水解的缩醛部位的聚乙二醇衍生物。记载了该聚乙二醇衍生物在生物体内被分解至适于从肾脏排出的分子量。然而,没有具体地促进从肾脏的排出的数据,进一步也没有有关细胞的空泡的记载。此外,已知这些能够水解的缩醛部位在血液中也缓慢分解,预计不能充分改善修饰后的生物相关物质的血液半衰期。
8.另一方面,存在为有效地释放药物而导入有分解性寡肽的聚乙二醇衍生物或在体内进行分解的水凝胶等的报道例子。
9.非专利文献3记载了具有通过酶而进行分解的寡肽部位的聚乙二醇衍生物。其中报道了,寡肽作为抗癌剂与聚乙二醇间的接头(linker)而被导入,通过在肿瘤周围特异性表达的酶,寡肽分解,可高效地释放抗癌剂。其目的在于抗癌剂的释放,并非出于抑制细胞空泡的目的而向聚乙二醇赋予分解性。
10.非专利文献4记载了使用具有通过酶进行分解的寡肽部位的交联分子和多分支型聚乙二醇衍生物的水凝胶。其中,寡肽被用作为连接多分支型聚乙二醇衍生物的交联分子,进一步地可以赋予水凝胶以经由酶的分解性。其目的在制备分解性的水凝胶,并非出于抑制细胞空泡的目的而向聚乙二醇赋予分解性。
11.专利文献3中记载了以寡肽作为骨架的分支型聚乙二醇衍生物。其中寡肽被用作为聚乙二醇衍生物的基本骨架,而非赋予经由酶的分解性。此外,其特征在于寡肽中包含赖氨酸、天冬氨酸等在侧链具有氨基或羧基的氨基酸,其目的在于合成将这些氨基酸利用于反应的分支型聚乙二醇衍生物。并不是以抑制细胞的空泡为目的的聚乙二醇衍生物。
12.进一步在用于对生物相关物质进行修饰的用途的聚乙二醇衍生物中,一般有直链型和分支型,非专利文献5中记载了分支型比直链型显著使生物相关物质的血液半衰期延长。上市的聚乙二醇改性制剂大多采用分支型。然而,迄今为止,在该领域还没有关于抑制细胞空泡的被分支型聚乙二醇衍生物修饰的生物相关物质的报道。
13.如上所述地,需要一种分支型的高分子量的聚乙二醇衍生物所修饰的生物相关物质,其在血液中稳定,可改善修饰的生物相关物质的血液半衰期,进一步可在细胞内特异性分解,抑制细胞的空泡的产生。现有技术文献专利文献
14.专利文献1:日本特表2009
‑
527581号公报专利文献2:国际公布第2005/108463号专利文献3:国际公布第2006/088248号非专利文献
15.非专利文献1:ema/chmp/swp/647258/2012非专利文献2:daniel g.rudmann,et al.,toxicol.pathol.,41,970
‑
983(2013)非专利文献3:francesco m veronese,et al.,bioconjugate chem.,16,775
‑
784(2005)非专利文献4:jiyuan yang,et al.,marcomol.biosci.,10(4),445
‑
454(2010)非专利文献5:yulia vugmeysterang,et al.,bioconjugate chem.,23,1452
‑
1462(2012)
技术实现要素:
发明所要解决的问题
16.本发明的问题在于提供一种键合有不引起细胞的空泡的高分子量的分支型聚乙二醇衍生物的生物相关物质。更具体地,在于提供一种生物相关物质,其在生物体内血液中稳定,并且被在细胞内分解的分解性聚乙二醇衍生物修饰,改善血液半衰期。用以解决问题的手段
17.本发明人为了解决上述问题而进行了深入研究,结果发明了具有键合有在细胞内进行分解的寡肽的分支型分解性聚乙二醇衍生物的生物相关物质。
18.即,本发明为如下所示。[1]一种生物相关物质,其键合有下式(a)所示的分解性聚乙二醇衍生物:
[0019]
[化1]
[0020]
(式中,n为45~950,w为以谷氨酸为中心的对称结构的5~47个残基的寡肽,a为2~8,d为生物相关物质,l1和l2分别独立地为2价间隔基团,以及b为1~40)。[2]一种生物相关物质,其键合有下式(1)所示的分解性聚乙二醇衍生物:
[0021]
[化2]
[0022]
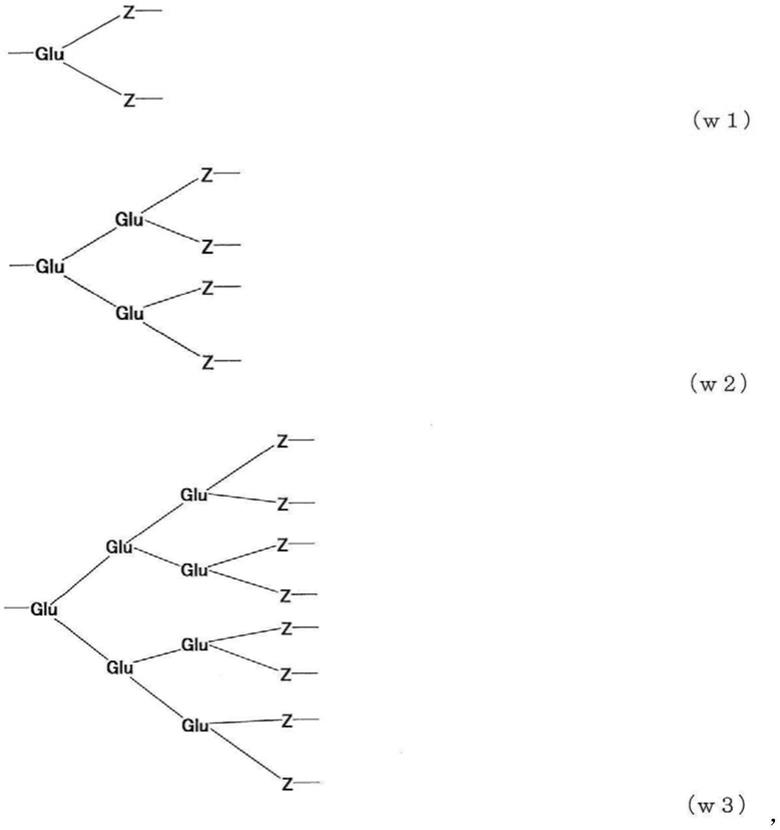
(式中,n为45~950,w为以谷氨酸为中心的对称结构的5~47个残基的寡肽,a为2~8,d为生物相关物质,以及l1和l2分别独立地为2价间隔基团)。[3]根据[1]或[2]所述的生物相关物质,其中,w的以谷氨酸为中心的对称结构的寡肽是具有以下w1、w2或w3的结构的寡肽。
[0023]
[化3]
[0024]
[化4]
[0025]
[化5]
[0026]
(式中,glu为谷氨酸的残基,以及z为由不包括半胱氨酸的中性氨基酸构成的2~5个残基的分解性寡肽。)[4]根据[3]所述的生物相关物质,其中,z的分解性寡肽是具有甘氨酸作为c末端的氨基酸的寡肽。[5]根据[3]或[4]中任一项所述的生物相关物质,其中,z的分解性寡肽是具有至少1个亲水指数为2.5以上的疏水性的中性氨基酸的寡肽。[6]根据[1]~[5]中任一项所述的生物相关物质,其中,分解性聚乙二醇衍生物的1分子的分子量为20,000以上。[7]根据[1]~[6]中任一项所述的生物相关物质,其中l1是氨基甲酸酯键、酰胺键、醚键、硫醚键、仲氨基、羰基、脲键、三唑基、马来酰亚胺与巯基的键、或肟键;或可包含这些键和/或基团的亚烷基。[8]根据[1]~[7]中任一项所述的生物相关物质,其中,l2是包含选自亚烷基;或氨基甲酸酯键、酰胺键、醚键、硫醚键、仲氨基、羰基和脲键中的至少一个键和/或基团的亚烷基。[9]根据[1]~[8]中任一项所述的生物相关物质,其中,d的生物相关物质为激素、细胞因子、抗体、适配体或酶。发明效果
[0027]
本发明的生物相关物质在生物体内的血液中稳定,由在结构内具有经由细胞内的酶分解的寡肽的分支型分解性聚乙二醇衍生物所修饰。因此,该生物相关物质在血液中稳定,具有与现有的不具有分解性的聚乙二醇衍生物所修饰的生物相关物质等同的血液半衰期。进一步地,该生物相关物质在被摄取至细胞内时,分解性聚乙二醇衍生物的寡肽部位迅
速被分解,因而可以抑制细胞空泡的产生这一悬而未决的问题。此外,构成分解性聚乙二醇衍生物的寡肽具有以谷氨酸为中心的对称结构,所有聚乙二醇链的末端键合有相同的分解性寡肽z。因此在细胞内分解时产生的聚乙二醇分解产物具有相同的分子量和相同的结构,具有从组织和细胞均匀排出的特点。
附图说明
[0028]
图1表示实施例1的化合物(p3)(nh2‑
e(fg
‑
200me)2)的gpc分析结果。图2表示在实施例8的使用细胞的分解性试验中,从细胞内回收的化合物(p3)(nh2‑
e(fg
‑
200me)2)的gpc分析结果。图3表示实施例5的化合物(p13)(nh2‑
e{e(fg
‑
100me)2}2)的gpc分析结果。图4表示在实施例8的使用细胞的分解性试验中,从细胞内回收的化合物(p13)(nh2‑
e{e(fg
‑
100me)2}2)的gpc分析结果。图5表示与实施例9的鲑降钙素的键合物(1)、甲氧基peg40kda
‑
sct的rplc分析结果。图6表示实施例3中所得的化合物(p8)和实施例9中所得的键合物(1)的maldi
‑
tof
‑
ms的分析结果。图7表示甲氧基peg醛40kda和实施例9中所得的甲氧基peg40kda
‑
sct的maldi
‑
tof
‑
ms的分析结果。图8表示与实施例9的鲑降钙素的键合物(1)、甲氧基peg40kda
‑
sct的sds
‑
page的分析结果(左图:cbb染色;右图:碘染色)。图9表示与实施例10的人生长激素的键合物(2)、甲氧基peg40kda
‑
hgh的rplc分析结果。图10表示与实施例10的人生长激素的键合物(2)的maldi
‑
tof
‑
ms的分析结果。图11表示与实施例10的人生长激素的甲氧基peg醛40kda
‑
hgh的maldi
‑
tof
‑
ms的分析结果。图12表示与实施例10的人生长激素的键合物(2)、甲氧基peg40kda
‑
hgh的sds
‑
page的分析结果(左图:cbb染色;右图:碘染色)。图13表示实施例13的长期给药甲氧基peg胺40kda的小鼠脑脉络丛的切片的图像(箭头指示空泡)。图14表示实施例13的长期给药化合物(p3)(nh2‑
e(fg
‑
200me)2)的小鼠脑脉络丛的切片的图像。图15表示实施例14的长期给药pbs、甲氧基peg胺40kda、甲氧基peg胺20kda、化合物(p3)(nh2‑
e(fg
‑
200me)2)的小鼠脑脉络丛的切片的图像(被染色的部分显示peg的蓄积)。图16表示实施例12的鲑降钙素和peg化鲑降钙素的生理活性(血液中钙浓度)的评价结果。
具体实施方式
[0029]
以下详细说明本发明。
[0030]
本发明所涉及的键合有分解性聚乙二醇衍生物的生物相关物质如下式(a)所示。
[0031]
[化6]
[0032]
(式中,n为45~950,w为以谷氨酸为中心的对称结构的5~47个残基的寡肽,a为2~8,d为生物相关物质,l1和l2分别独立地为2价间隔基团,以及b为1~40。)
[0033]
与本发明的式(a)的生物相关物质键合的聚乙二醇衍生物1分子的分子量通常为4,000~160,000,优选为10,000~120,000,进一步优选为20,000~80,000。本发明的一个优选的实施方式中,本发明的式(a)的聚乙二醇衍生物1分子的分子量为20,000以上。此处所称的分子量是指数均分子量(mn)。
[0034]
式(a)中的n为聚乙二醇的重复单元数,通常为45~950,优选为110~690,进一步优选为220~460。
[0035]
式(a)中的a为与寡肽键合的聚乙二醇链的根数,通常为2~8,优选为2或4或8,进一步优选为2或4。
[0036]
式(a)中的b是与生物相关物质键合的分解性聚乙二醇衍生物的分子数,通常为1~40,优选为1~20,进一步优选为1~10。如果增加与生物相关物质键合的聚乙二醇衍生物的分子数,则获得血液半衰期延长、抗原性降低等效果,但存在因生物相关物质而使活性降低的可能性。另一方面,已知在一部分的酶等生物相关物质中,即使键合多个聚乙二醇衍生物,活性也不降低。
[0037]
式(a)中的l1和l2分别独立地为2价间隔基团,这些间隔基团只要是能够形成共价键的基团,就没有特别限制,l1优选为酰胺键、醚键、硫醚键、氨基甲酸酯键、仲氨基、羰基、脲键、三唑基、马来酰亚胺与巯基的键、或肟键;或可包含这些键和/或基团的亚烷基。另外,l2优选为亚烷基;或包含选自酰胺键、醚键、硫醚键、氨基甲酸酯键、仲氨基、羰基和脲键中的至少一个键和/或基团的亚烷基。l2优选以碳原子键合于聚乙二醇的重复单元。l1和l2的特别优选方式如下述的群组(i)所示。此外,也可以将群组(i)的间隔基团组合2个至5个。作为2价间隔基团,酯键和碳酸酯键由于在生物体内血液中缓慢分解因而并不合适。
[0038]
群组(i):
[0039]
[化7]
[0040]
(z1)~(z20)中,式中的s表示0~10的整数,优选表示0~6的整数,进一步优选表示0~3的整数。此外,(z2)~(z20)中,式中的s可相同,也可不同。l1为非对称性2价间隔基团时,与相邻的其他基团的键合位置并没有特别限定,如果上述群组(i)的由上式表示的间隔基团的右侧表示与w的键合位置,左侧表示与d的键合位置时,则可采取左侧表示与w的键合位置、右侧表示与d的键合位置时的两个键合位置。同样地,l2为非对称性2价间隔基团时,如果上述群组(i)的由上式表示的间隔基团的右侧表示与och2ch2的键合位置、左侧表示与w的键合位置时,则可采取左侧表示与och2ch2的键合位置、右侧表示与w的键合位置时的两个键合位置。
[0041]
作为式(a)中的l1,优选群组(i)的(z3)、(z6)、(z7)~(z20)所示的基团,更优选(z6)、(z9)、(z10)、(z12)、(z14)、(z16)、(z18)或(z20)所示的基团,进一步优选(z10)、(z12)、(z16)或(z20)所示的基团。作为式(a)中的l2,优选群组(i)的(z1)、(z2)、(z3)、(z4)、(z5)、(z6)、(z7)或(z8)所示的基团,更优选(z3)或(z5)所示的基团。
[0042]
式(a)中的w只要是以谷氨酸为中心的对称结构的5~47个残基的寡肽,在生物体内血液中稳定,且经细胞内的酶进行分解的寡肽,就没有特别限制,作为构成寡肽的氨基酸,除了构成中心部分的谷氨酸以外,优选由不包括半胱氨酸的中性氨基酸构成。此处所称的以谷氨酸为中心的对称结构的寡肽是指在谷氨酸的α位的羧基与γ位的羧基上键合有相同肽的化合物,以谷氨酸为中心成对的肽是采用对称结构的寡肽。作为该寡肽中的中性氨基酸与谷氨酸数目的构成比(中性氨基酸的数目/谷氨酸的数目),通常为2~10,优选为2~8,进一步优选为2~6。构成w的氨基酸基本上为l型。
[0043]
w的特别优选方式如下述的群组(ii)所示。
[0044]
群组(ii):
[0045]
[化8]
[0046]
[化9]
[0047]
[化10]
[0048]
(式中,glu为谷氨酸的残基,以及z为由不包括半胱氨酸的中性氨基酸构成的2~5个残基的分解性寡肽。)
[0049]
(w1)~(w3)中的z优选为在侧链上具有氨基或羧基的氨基酸,具体地由不含赖氨酸、天冬氨酸、或谷氨酸的中性氨基酸构成的寡肽。本发明的式(a)的分支型分解性聚乙二醇衍生物的合成中,在通过反应键合作为原料的聚乙二醇衍生物与寡肽时,将寡肽的c末端
的羧基利用于与聚乙二醇衍生物的缩合反应中。然而,该寡肽具有在侧链上具有氨基或羧基的氨基酸时,通过缩合反应发生寡肽之间的副反应、聚乙二醇衍生物不被导入至作为目标的c末端的羧基而导入至侧链的羧基的杂质。由于该杂质难以通过通常的萃取、析晶等提纯工序去除,因而为获得纯度良好的目标物,理想的是使用由侧链上不具有氨基或羧基的氨基酸构成的寡肽。构成z的氨基酸为α
‑
氨基酸,此外基本上为l型。
[0050]
由于作为中性氨基酸的半胱氨酸具有巯基,与其他巯基形成双硫键,(w1)~(w3)中的z优选为由不包括半胱氨酸的中性氨基酸构成的寡肽。
[0051]
而且,(w1)~(w3)中的z优选为具有甘氨酸作为c末端的氨基酸的寡肽。在使c末端的羧基与聚乙二醇衍生物进行反应时,基本上需要用缩合剂等使c末端的羧基活性化。已知的是在该活性化的工序中,甘氨酸以外的氨基酸易于发生差向异构化,生成副产物立体异构体。通过使寡肽的c末端的氨基酸为非手性的甘氨酸,可以得到不会有立体异构体副产出来的高纯度的目标物。
[0052]
进一步地,(w1)~(w3)中的z是亲水指数为2.5以上的疏水性的中性氨基酸,具体地优选为具有至少1个苯丙氨酸、亮氨酸、缬氨酸、异亮氨酸的寡肽,进一步优选为具有苯丙氨酸的寡肽。由凯特(kyte)和杜利特尔(doolittle)所完成的定量表示氨基酸的疏水性的亲水指数(hydropathy index),其值越大,表示越为疏水性氨基酸(kyte j&doolittle rf,1982,j mol biol,157:105
‑
132.)。
[0053]
(w1)~(w3)中的z只要是在生物体内血液中稳定,且具有经细胞内的酶进行分解的性能,由不包括半胱氨酸的中性氨基酸构成的2~5个残基的寡肽,就没有特别限制,作为具体的例子,为甘氨酸
‑
苯丙氨酸
‑
亮氨酸
‑
甘氨酸、甘氨酸
‑
甘氨酸
‑
苯丙氨酸
‑
甘氨酸、甘氨酸
‑
苯丙氨酸
‑
甘氨酸、甘氨酸
‑
亮氨酸
‑
甘氨酸、缬氨酸
‑
瓜氨酸
‑
甘氨酸、缬氨酸
‑
丙氨酸
‑
甘氨酸、苯丙氨酸
‑
甘氨酸等,优选为甘氨酸
‑
苯丙氨酸
‑
亮氨酸
‑
甘氨酸、甘氨酸
‑
甘氨酸
‑
苯丙氨酸
‑
甘氨酸、甘氨酸
‑
苯丙氨酸
‑
甘氨酸、缬氨酸
‑
瓜氨酸
‑
甘氨酸、缬氨酸
‑
丙氨酸
‑
甘氨酸或苯丙氨酸
‑
甘氨酸,更优选为甘氨酸
‑
苯丙氨酸
‑
亮氨酸
‑
甘氨酸、甘氨酸
‑
苯丙氨酸
‑
甘氨酸、缬氨酸
‑
瓜氨酸
‑
甘氨酸或苯丙氨酸
‑
甘氨酸,进一步更优选为甘氨酸
‑
苯丙氨酸
‑
亮氨酸
‑
甘氨酸或苯丙氨酸
‑
甘氨酸。
[0054]
式(a)中的d为生物相关物质,没有特别限制,但是与人或其他动物的疾病的诊断、治愈、缓解、治疗或预防有关的物质。具体地包括蛋白质、肽、核酸、细胞、病毒等,作为适宜的蛋白质或肽,可举出激素、细胞因子、抗体、适配体、酶等。更具体地,作为细胞因子,可举出调节免疫的干扰素i型、ii型、iii型和白细胞介素、肿瘤坏死因子、它们的受体拮抗剂等。作为生长因子,可举出作为造血生长因子的促红细胞生成素、作为刺激因子的粒细胞集落刺激因子(gcsf)等,作为血液凝固因子,可举出第v因子、第vii因子、第viii因子、第ix因子、第x因子、第xii因子等。作为激素,可举出降钙素、胰岛素、其类似物和艾塞那肽(exenatide)、glp
‑
1,以及生长抑素、人生长激素等。作为抗体,可举出完全抗体,此外作为抗体片段,可举出fab、svfv等,作为适配体,可举出dna适配体、rna适配体等,作为酶,可举出超氧化物歧化酶、尿酸酶等。理想的是这些蛋白质在血液中的稳定性低,以聚乙二醇进行修饰,延长血液半衰期。作为适宜的蛋白质,可举出干扰素、白细胞介素、促红细胞生成素、gcsf、第viii因
子、第ix因子、人生长激素、抗体片段等,更优选地可举出人生长激素、干扰素、gcsf、促红细胞生成素或抗体片段(特别是fab),进一步优选地可举出人生长激素或gcsf。作为适宜的肽,可举出胰岛素、比伐卢定(bivalirudin)、特立帕肽(teriparatide)、艾塞那肽(exenatide)、恩夫韦肽(enfuvirtide)、地加瑞克(degarelix)、米法莫肽(mifamurtide)、奈西利肽(nesiritide)、戈舍瑞林(goserelin)、格拉替雷(glatiramer)、奥曲肽(octreotide)、兰瑞肽(lanreotide)、艾替班特(icatibant)、齐考诺肽(ziconotide、普兰林肽(pramlintide)、罗米司亭(romiplostim)、降钙素、催产素、亮丙瑞林(leuprorelin)、胰高血糖素,更优选为地可举出胰岛素、艾塞那肽、降钙素(特别是鲑降钙素)。
[0055]
式(a)所示的生物相关物质的优选方式之一是,b为1的下式(1)所示的生物相关物质。
[0056]
[化11]
[0057]
(式中,n、w、a、d、l1和l2分别与上述含义相同。)
[0058]
式(1)的优选方式之一是,w为w1和a=2的下式(2)所示的生物相关物质。
[0059]
[化12]
[0060]
(式中,glu、z、n、d、l1和l2与上述含义相同。)
[0061]
式(1)的优选方式之一是,w为w2以及a=4的下式(3)所示的生物相关物质。
[0062]
[化13]
[0063]
(式中,glu、z、n、d、l1和l2与上述含义相同。)
[0064]
式(1)的优选方式之一是,w为w3以及a=8的下式(4)所示的生物相关物质。
[0065]
[化14]
[0066]
(式中,glu、z、n、d、l1和l2与上述含义相同。)
[0067]
本发明的式(a)的生物相关物质可以通过使下式(5)所示的分解性聚乙二醇衍生物与生物相关物质进行反应而获得。
[0068]
[化15]
[0069]
(式中,x为能够与生物相关物质反应的官能团,w、a、n、l1和l2与上述含义相同。)
[0070]
式(5)中的x只要是与存在于作为化学修饰对象的生理活性蛋白质、肽、抗体、核酸等生物相关物质中的官能团进行反应而形成共价键的官能团,就没有特别限制。例如,可举出“harris,j.m.poly(ethylene glycol)chemistry;plenum press:new york,1992(j.m.哈里斯,聚乙二醇化学;普莱南出版社:纽约,1992)”、“hermanson,g.t.bioconjugate techniques,2nd ed.;academic press:san diego,ca,2008(g.t.赫曼森,生物偶联技术,第二版;学术出版社:加利福尼亚州圣地亚哥,2008)”和“pegylated protein drugs:basic science and clinical applications;veronese,f.m.,ed.;birkhauser:basel,switzerland,2009(聚乙二醇化蛋白药物:基础科学和临床应用,f.m.韦罗内塞编;博克豪斯出版社:瑞士巴塞尔,2009)”等所记载的官能团。
[0071]
式(5)中的x所示的“能够与生物相关物质反应的官能团”,只要是能够与生物相关物质所具有的氨基、巯基、醛基、羧基、不饱和键或叠氮基等官能团化学键合的官能团,就没有特别限制。具体可举出活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧化物基、羧基、巯基、马来酰亚胺基、取代马来酰亚胺基、酰肼基、二硫代吡啶基、取代磺酸酯基、乙烯基磺酰基、氨基、氧氨基(h2n
‑
o
‑
基)、碘乙酰胺基、烷基羰基、烯基(例如烯丙基、乙烯基)、炔基、取代炔基(例如后述的被碳原子数1~5的烃基取代的炔基)、叠氮基、丙烯酸基、
磺酰氧基(例如烷基磺酰氧基)、α
‑
卤代乙酰基等,优选为活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧化物基、马来酰亚胺基、取代马来酰亚胺基、乙烯基磺酰基、丙烯酸基、磺酰氧基(例如碳原子数1~5的烷基
‑
磺酰氧基)、取代磺酸酯基、羧基、巯基、二硫代吡啶基、α
‑
卤代乙酰基、炔基、取代炔基(例如后述的被碳原子数1~5的烃基取代的碳原子数2~5的炔基)、烯丙基、乙烯基、氨基、氧氨基、酰肼基和叠氮基,更优选为活性酯基、活性碳酸酯基、醛基、马来酰亚胺基、氧氨基和氨基,特别优选为醛基、马来酰亚胺基和氧氨基。
[0072]
其他适宜的实施方式中,所述官能团x可分类为下述的群组(iii)、群组(iv)、群组(v)、群组(vi)、群组(vii)和群组(viii)。
[0073]
群组(iii):能够与生物相关物质所具有的氨基反应的官能团可举出下述的(a)、(b)、(c)、(d)、(e)、(f)、(g)、(j)或(k)所示的基团。
[0074]
群组(iv):能够与生物相关物质所具有的巯基反应的官能团可举出下述的(a)、(b)、(c)、(d)、(e)、(f)、(g)、(h)、(i)、(j)、(k)或(l)所示的基团。
[0075]
群组(v):能够与生物相关物质所具有的醛基反应的官能团可举出下述的(h)、(m)、(n)或(p)所示的基团。
[0076]
群组(vi):能够与生物相关物质所具有的羧基反应的官能团可举出下述的(h)、(m)、(n)或(p)所示的基团。
[0077]
群组(vii):能够与生物相关物质所具有的不饱和键反应的官能团可举出下述的(h)、(m)或(o)所示的基团。
[0078]
群组(viii):能够与生物相关物质所具有的叠氮基反应的官能团可举出下述的(l)所示的基团。
[0079]
[化16]
[0080]
官能团(j)中,式中的w1表示氯原子(cl)、溴原子(br)或碘原子(i)等卤素原子,优选为br或i,更优选为i。
[0081]
另外,官能团(e)和官能团(l)中,式中的y1和y3分别独立地表示氢原子或碳原子数1~5的烃基,优选为碳原子数1~5的烃基。作为碳原子数1~5的烃基,具体地可举出甲基、乙基、丙基、异丙基、丁基、叔丁基等,优选为甲基或乙基。
[0082]
另外,官能团(k)中,式中的y2表示可含有氟原子的碳原子数为1~10的烃基,具体地可举出甲基、乙基、丙基、异丙基、丁基、叔丁基、己基、壬基、乙烯基、苯基、苄基、4
‑
甲基苯基、三氟甲基、2,2,2
‑
三氟乙基、4
‑
(三氟甲氧基)苯基等,优选为甲基、乙烯基、4
‑
甲基苯基或2,2,2
‑
三氟乙基。
[0083]
活性酯基是指具有离去能力高的烷氧基的酯基。作为离去能力高的烷氧基,可举出衍生自硝基苯酚、n
‑
羟基琥珀酰亚胺、五氟苯酚等的烷氧基。活性酯基优选为具有衍生自n
‑
羟基琥珀酰亚胺的烷氧基的酯基。
[0084]
活性碳酸酯基是指具有离去能力高的烷氧基的碳酸酯基。作为离去能力高的烷氧基,可举出衍生自硝基苯酚、n
‑
羟基琥珀酰亚胺、五氟苯酚等的烷氧基。活性碳酸酯基优选为具有衍生自硝基苯酚或n
‑
羟基琥珀酰亚胺的烷氧基的碳酸酯基。
[0085]
取代马来酰亚胺基是指在马来酰亚胺基的双键之一的碳原子上键合有烃基的马来酰亚胺基。烃基具体地可举出甲基、乙基、丙基、异丙基、丁基、叔丁基等,优选为甲基或乙基。
[0086]
取代磺酸酯基是指在磺酸酯基的硫黄原子上键合有可含有氟原子的烃基的磺酸酯基。作为可含有氟原子的烃基,具体地可举出甲基、乙基、丙基、异丙基、丁基、叔丁基、己基、壬基、乙烯基、苯基、苄基、4
‑
甲基苯基、三氟甲基、2,2,2
‑
三氟乙基、4
‑
(三氟甲氧基)苯基等,优选为甲基、乙烯基、4
‑
甲基苯基或2,2,2
‑
三氟乙基。
[0087]
用于本发明的生物相关物质的分支型分解性聚乙二醇衍生物例如可以经过以下工序来制造。
[0088]
[化17]
[0089]
(工序中的peg为聚乙二醇链,肽为寡肽,pro为保护基,以及l3为2价间隔基团。)
[0090]
工序中的peg为聚乙二醇链,分子量如作为上述的聚乙二醇的重复单元数的n所定义地,即由于n为45~950,其分子量的范围为2000~42000。
[0091]
工序中的肽是与上述z含义相同的寡肽。本工序中使用n末端的氨基被保护基所保护的寡肽。
[0092]
工序中的pro为保护基,此处保护基是指防止或阻止某一反应条件下分子中的能够进行特定化学反应的官能团的反应的成分。保护基根据所保护的能够进行化学反应的官能团的种类、所使用的条件和分子中的其他官能团或保护基的存在而有所改变。保护基的具体例子可以在很多通常的书籍中找到,例如“wuts,p.g.m.;greene,t.w.protective groups in organic synthesis,4th ed.;wiley
‑
interscience:new york,2007(p.g.m.伍斯;t.w.格林,有机合成中的保护基,第四版;wiley
‑
interscience:纽约,2007)”所记载。此外,被保护基保护的官能团通过使用适宜各自保护基的反应条件进行脱保护、即进行化学反应,可再次产生原有的官能团。保护基的典型的脱保护条件如上述文献中所记载。
[0093]
工序中的l3是与上述l1和l2相同含义的2价间隔基团。
[0094]
反应a是通过缩合反应将n末端的氨基被保护基所保护的寡肽的羧基与单个末端为甲氧基的聚乙二醇衍生物的氨基键合而获得聚乙二醇衍生物(1)的工序。寡肽的n末端的氨基的保护基没有特别限制,例如可举出酰基系保护基和氨基甲酸酯系保护基,具体地可举出三氟乙酰基、9
‑
芴基甲氧基羰基(fmoc)、叔丁氧基羰基等。作为缩合反应,没有特别限制,但理想的是使用缩合剂的反应。作为缩合剂,可单独使用二环己基碳二亚胺(dcc)、1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳二亚胺盐酸盐(edc)等碳二亚胺系的缩合剂,也可与n
‑
羟基琥珀酰亚胺(nhs)、1
‑
羟基苯并三唑(hobt)、1
‑
羟基
‑7‑
氮杂苯并三唑(hoat)等试剂一起使用。此外,也可使用反应性更高的hatu或hbtu、tatu、tbtu、comu、dmt
‑
mm等缩合剂。此外为了促进反应,也可使用三乙胺或二甲基氨基吡啶等碱。反应中副反应生成的杂质或反应中未被消耗而残留的寡肽和缩合剂等优选进行
提纯而去除。提纯没有特别限制,能够以萃取、重结晶、吸附处理、再沉淀、柱色谱法、超临界萃取等进行提纯。
[0095]
[化18]
[0096]
脱保护b是将反应a中所得的聚乙二醇衍生物(1)的保护基进行脱保护而获得聚乙二醇衍生物(2)的工序。作为脱保护反应,可以使用现有公知的方法,但需要使用寡肽和l3的2价间隔基团不分解的条件。此外,本工序也可以作为反应a的工序的一部分来实施。脱保护反应中副反应生成的杂质等优选进行提纯而去除。提纯没有特别限制,能够以萃取、重结晶、吸附处理、再沉淀、柱色谱法、超临界萃取等进行提纯。
[0097]
[化19]
[0098]
反应c是通过缩合反应使脱保护b中所得的聚乙二醇衍生物(2)的氨基与氨基被保护基所保护的谷氨酸衍生物的两个羧基键合,从而获得2根分解性聚乙二醇链被谷氨酸残基连接的结构的分支型聚乙二醇衍生物(3)的工序。与上述反应a同样地,理想的是使用缩合剂的反应,为了促进反应,也可使用三乙胺或二甲基氨基吡啶等碱。谷氨酸的氨基的保护基没有特别限制,例如可举出酰基系保护基和氨基甲酸酯系保护基,具体地可举出三氟乙酰基、9
‑
芴基甲氧基羰基(fmoc)、叔丁氧基羰基等。反应中副反应生成的杂质或反应中未被消耗而残留的聚乙二醇衍生物等优选进行提纯而去除。提纯没有特别限制,能够以萃取、重结晶、吸附处理、再沉淀、柱色谱法、超临界萃取等进行提纯。
[0099]
[化20]
[0100]
脱保护d是将反应c中所得的聚乙二醇衍生物(3)的保护基脱保护而获得聚乙二醇衍生物(4)的工序。作为脱保护反应,可以使用现有公知的方法,但需要使用寡肽和l3的2价间隔基团不分解的条件。此外,本工序也可以作为反应c的工序的一部分来实施。脱保护反应中副反应生成的杂质等优选进行提纯而去除。提纯没有特别限制,能够以萃取、重结晶、吸附处理、再沉淀、柱色谱法、超临界萃取等进行提纯。
[0101]
[化21]
[0102]
反应e是通过缩合反应使脱保护d中所得的聚乙二醇衍生物(4)的氨基与氨基被保护基所保护的谷氨酸衍生物的两个羧基键合,从而获得4根分解性聚乙二醇链被谷氨酸残基连接的结构的分支型聚乙二醇衍生物(5)的工序。可以在与上述反应c相同条件下反应和提纯。作为从聚乙二醇衍生物(5)中除去子量或官能团不同的聚乙二醇杂质的方法,可以使用日本专利特开2014
‑
208786号公报或日本专利特开2011
‑
79934号公报中记载的提纯技术。
[0103]
[化22]
[0104]
脱保护f是将反应e中所得的聚乙二醇衍生物(5)的保护基脱保护而获得聚乙二醇衍生物(6)的工序。作为脱保护反应,可以使用现有公知的方法,但需要使用寡肽和l3的2价间隔基团不分解的条件。能够在与上述脱保护d相同条件下反应和提纯。此外,本工序也可以作为反应e的工序的一部分来实施。
[0105]
[化23]
[0106]
反应g是通过缩合反应使脱保护f中所得的聚乙二醇衍生物(6)的氨基与氨基被保护基所保护的谷氨酸衍生物的两个羧基键合,从而获得8根分解性聚乙二醇链被谷氨酸残基连接的结构的分支型聚乙二醇衍生物(7)的工序。可以在与上述反应c相同条件下反应和提纯。
[0107]
[化24]
[0108]
脱保护h是将反应g中所得的聚乙二醇衍生物(7)的保护基脱保护而获得聚乙二醇衍生物(8)的工序。能够在与上述脱保护f相同条件下反应和提纯。此外,本工序也可以作为反应g的工序的一部分来实施。
[0109]
通过进行以上的反应a、脱保护b、反应c和脱保护d,可得到2分支型分解性聚乙二醇衍生物(4)。将2分支型分解性聚乙二醇衍生物(4)作为原料,继续通过进行反应e和脱保护f,可得到4分支型分解性聚乙二醇衍生物(6)。进一步继续通过进行反应g和脱保护h,可得到8分支型分解性聚乙二醇衍生物(8)。
[0110]
脱保护d、脱保护f和脱保护h中所得的聚乙二醇衍生物(4)、(6)和(8)均具有一个氨基,可利用其转换为各种官能团。
[0111]
关于将聚乙二醇衍生物的末端的氨基转换为其他官能团的工序,没有特别限制,基本上可以通过使用具有能够与氨基反应的活性酯基的化合物或酸酐、酰氯等通常的反应试剂,容易地转换为各种官能团。
[0112]
例如,在希望将聚乙二醇衍生物的末端的氨基转换为马来酰亚胺基时,可以通过使其与如下的试剂进行反应而获得目标物。
[0113]
[化25]
[0114]
例如,在希望将聚乙二醇衍生物的末端的氨基转换为羧基时,可以通过使其与琥珀酸酐或戊二酸酐进行反应而获得目标物。
[0115]
例如,在希望将聚乙二醇衍生物的末端的氨基转换为水酸基时,通过与己内酯等环状酯的开环物进行缩合反应而获得目标物。
[0116]
这些反应试剂是低分子量的试剂,由于与高分子量聚合物的聚乙二醇衍生物有很大的溶解性差别,因此可以通过萃取或析晶等通常的提纯方法容易地去除。
[0117]
要求经由如上所述的工序而获得的分解性聚乙二醇在血液中稳定,具有仅在细胞内进行分解的性能。为了恰当地评价其性能,例如可以进行如下所示的试验,评价分解性聚乙二醇在血液中的稳定性,以及在细胞内的分解性。此外,在这些评价中,考虑聚乙二醇衍生物所具有的官能团种类引起的影响,评价试样全部对具有一个氨基的聚乙二醇衍生物统一进行了试验。
[0118]
关于用于评价分解性聚乙二醇衍生物在血液中的稳定性的试验方法,没有特别限制,例如可举出使用小鼠、大鼠、人等血清的试验等。具体地,可以通过在血清中溶解聚乙二醇衍生物至浓度成为1~10mg/ml,在37℃下孵育96小时后,回收血清中所含的聚乙二醇衍生物,测定gpc,从而评价分解率。分解率基于稳定性试验前的聚乙二醇衍生物的gpc主馏分的峰面积%和稳定性试验后的聚乙二醇衍生物的gpc主馏分的峰面积%进行计算。具体地使用下式。分解率=(试验前的峰面积%
‑
试验后的峰面积%)
÷
试验前的峰面积%
×
100例如,如果稳定性试验前的分解性聚乙二醇衍生物的gpc主馏分的峰面积%为95%,试验后的gpc主馏分的峰面积%变为90%时,分解率如下地进行计算。分解率=(95
‑
90)
÷
95
×
100=5.26(%)分解性聚乙二醇衍生物一旦在血液中分解,则不能获得目标的血液半衰期,因此在稳定性试验中,96小时后的分解率优选10%以下,进一步优选5%以下。
[0119]
关于用于评价分解性聚乙二醇衍生物在细胞内的分解性的试验方法,没有特别限制,例如可举出使用含有分解性聚乙二醇衍生物的培养基,培养细胞的试验等。关于此处使用的细胞或培养基,没有特别限制,具体地,可以通过在培养基rpmi
‑
1640中溶解聚乙二醇衍生物至浓度成为1~20mg/ml,使用该培养基,在37℃下培养96小时巨噬细胞raw264.7后,回收细胞中的聚乙二醇衍生物,测定gpc,从而评价分解率。分解率与稳定性试验同样地,可以使用试验前后的聚乙二醇衍生物的gpc主馏分的峰面积%进行计算。例如,如果使用细胞的分解性试验前的分解性聚乙二醇衍生物的gpc主馏分的峰面积%为95%,试验后的gpc主馏分的峰面积%变为5%时,分解率如下地进行计算。分解率=(95
‑
5)
÷
95
×
100=94.7(%)如果分解性聚乙二醇衍生物不能在细胞内高效地分解,则不能抑制作为目标的细胞的空泡,因此在分解性试验中,96小时后的分解率优选90%以上,进一步优选95%以上。
[0120]
作为使获得的分解性聚乙二醇衍生物与生物相关物质键合的方法,没有特别限制,例如可以使用“hermanson,g.t.bioconjugate techniques,3rd ed.;academic press:san diego,ca,2013(g.t.赫曼森,生物偶联技术,第三版;学术出版社:加利福尼亚州圣地亚哥,2013)”、“mark,sonny s.bioconjugate protocols,strategies and methods;2011(马克,桑尼s.,生物偶联方案、策略和方法;2011)”中记载的方法。这些之中,例如,在将作为生物相关物质的蛋白质、肽等赖氨酸残基的侧链氨基作为标靶(target)时,使用具有活化酯基、活化碳酸酯基的聚乙二醇衍生物。此外,以作为生物相关物质的蛋白质、肽等半胱
氨酸残基的巯基作为标靶时,使用具有马来酰亚胺基、碘乙酰胺基的聚乙二醇衍生物。由于天然的生物相关物质中所含的游离半胱氨酸残基的数量极少,因此用该方法可进一步选择性地使聚乙二醇与生物相关物质键合。进一步地,作为产生或导入硫醇基的方法,有切断生物相关物质的双硫键的方法、通过基因工程改变生物相关物质而导入半胱氨酸残基的方法等,已知的是通过与这些技术相组合,可以在生物相关物质的目标部位键合目标个数的聚乙二醇衍生物。接着,在以作为生物相关物质的蛋白质、肽等n末端的氨基作为标靶时,使用具有醛基的聚乙二醇衍生物。具体地,通过在低ph的缓冲溶液中,使用具有醛基的聚乙二醇衍生物和合适的还原剂,可以使聚乙二醇衍生物选择性地键合在蛋白质、肽的n末端的氨基上。这些键合有聚乙二醇衍生物的生物相关物质可以通过作为通常方法而周知的透析、凝胶渗透色谱法(gpc)、离子交换色谱法(iec)等进行提纯。此外,获得的生物相关物质通常可以用基质辅助激光解吸电离飞行时间质谱仪(maldi
‑
tof
‑
ms)、聚丙烯酰胺凝胶电泳(sds
‑
page)、反相色谱法(rplc)等分析方法进行评价。
[0121]
关于评价键合有分解性聚乙二醇衍生物的生物相关物质的生理活性的方法,没有特别限制,例如,如果生物相关物质为胰岛素则测定血液中糖浓度,如果为降钙素则测定血液中钙浓度等,可以通过从给药后的动物进行定期采血,用合适的分析仪器测定血液中的物质而进行评价。具体地,如果为胰岛素,则可以使用葡萄糖测定试剂盒,监测给药后的葡萄糖浓度的降低,如果为降钙素,则可以使用钙测定试剂盒,监测给药后的钙浓度的降低而进行评价。
[0122]
关于用于评价键合有分解性聚乙二醇衍生物的生物相关物质的血液半衰期和体内分布的试验方法,没有特别限制,例如可举出用放射性同位素或荧光物质标记,对小鼠或大鼠给药,并进行监测的试验等。认为导入至聚乙二醇衍生物的分解性肽赋予聚乙二醇以在细胞内的分解性,但因该肽结构而存在改变键合有聚乙二醇的生物相关物质的体内动力学的可能性。因此,为了确认导入的肽结构对体内动力学的影响,对于血液半衰期及其体内分布,有必要与修饰了不具有分解性的同分子量的聚乙二醇衍生物的生物相关物质进行比较。具体地,可以在以放射性同位素标记后的生物相关物质上,分别键合不具有分解性的聚乙二醇衍生物和分解性聚乙二醇衍生物,将获得的2种生物相关物质向小鼠给药,在多个时间点测定血液、各脏器的辐射剂量,进行定量测定。
[0123]
关于用于评价分解性聚乙二醇衍生物的血液半衰期和体内分布的试验方法,没有特别限制,例如可举出用放射性同位素或荧光物质标记,对小鼠或大鼠给药,并进行监测的试验等。认为导入至聚乙二醇衍生物的分解性肽对聚乙二醇赋予在细胞内的分解性,但因该肽结构而存在改变聚乙二醇的药代动力学的可能性。因此,为了确认导入的肽结构对药代动力学的影响,对于血液半衰期及其体内分布,需要与不具有分解性的相同分子量的聚乙二醇衍生物进行比较。具体地,可以将用放射性同位素标记的不具有分解性的聚乙二醇衍生物和分解性聚乙二醇衍生物向小鼠给药,在多个时间点测定血液、各脏器的辐射剂量,进行定量测定。
[0124]
关于用于评价分解性聚乙二醇衍生物的细胞的空泡抑制的试验方法,没有特别限
制,例如可举出如非专利文献2所记载地,持续长期间、高频度、高给药量地向小鼠或大鼠给药,对被认为易于产生空泡的脏器或器官的切片图像进行确认的试验等。具体地,可在生理盐水中溶解聚乙二醇衍生物至浓度成为10~250mg/ml,通过小鼠尾静脉,持续每周3次、4周以上、20~100μl的给药,制作被认为易于产生空泡的器官的脑脉络丛或脾脏等的石蜡切片进行染色后,通过病理学方法确认切片图像,进行空泡抑制的评价。此外,本评价中聚乙二醇的给药量相比于该技术领域中的通常的聚乙二醇的给药量,需要给予过量很大的聚乙二醇。
[0125]
非专利文献2中有高分子量的聚乙二醇引起的细胞的空泡化与聚乙二醇在组织中的蓄积有关这样的记载。关于用于评价分解性聚乙二醇衍生物的细胞中的蓄积性的试验方法,没有特别限制,可以通过与以上述空泡评价相同方法制作的切片图像进行评价。可以通过病理学方法确认被认为易于产生空泡的器官的脑脉络丛或脾脏等的染色后的切片图像,进行聚乙二醇的蓄积性的评价。此外,本评价中聚乙二醇的给药量相比于该技术领域中的通常的聚乙二醇的给药量,需要给予过量很大的聚乙二醇。实施例
[0126]
下述实施例中获得的1h
‑
nmr得自捷欧迪拓姆(jeoldatum)株式会社制造的jnm
‑
ecp400或jnm
‑
eca600。测定中使用样品管,氘代溶剂中使用d2o或含有四甲基硅烷(tms)作为内标物的cdcl3和d6‑
dmso。获得的聚乙二醇衍生物的分子量和胺纯度使用液相色谱(gpc和hplc)进行计算。液相色谱的系统是在gpc中使用东曹株式会社制造的“hlc
‑
8320gpc ecosec”,在hplc中使用waters公司制造的“alliance”。以下示出gpc和hplc的分析条件。gpc分析(分子量测定)标准聚合物:使用分子量为8,000、20,000、50,000和100,000的聚乙二醇作为标准聚合物进行利用gpc分析的分子量测定。检测器:示差折光仪色谱柱:ultrahydrogel 500和ultrahydrogel 250(waters公司制造)流动相:100mm乙酸盐缓冲液(acetate buffer) 0.02%nan3(ph5.2)流速:0.5ml/min样品量:5mg/ml,20μl色谱柱温度:30℃hplc分析(胺纯度测定)检测器:示差折光仪色谱柱:tskgel sp
‑
5pw(东曹株式会社制造)流动相:1mm磷酸钠缓冲液(sodium phosphate buffer)(ph6.5)流速:0.5ml/min注入量:5mg/ml,20μl色谱柱温度:40℃
[0127]
[实施例1]
化合物(p3)(nh2‑
e(fg
‑
200me)2)的合成
[0128]
[化26]
[0129]
[实施例1
‑
1]
[0130]
[化27]
[0131]
向n末端被9
‑
芴基甲氧基羰基(fmoc基)保护的l
‑
苯丙氨酰
‑
甘氨酸(fmoc
‑
phe
‑
gly)(0.267g,6.0
×
10
‑4mol,渡边化学工业株式会社制造)和末端具有丙基氨基的甲氧基peg(6.0g,2.8
×
10
‑4mol,数均分子量=21,120,日油株式会社制造的“sunbright mepa
‑
20t”)中添加脱水n,n
’‑
二甲基甲酰胺(60g),在30℃下加温溶解。然后,添加二异丙基乙胺(192μl,1.2
×
10
‑3mol,关东化学株式会社制造)和(1
‑
氰基
‑2‑
乙氧基
‑2‑
氧代亚乙基氨基氧基)二甲基氨基
‑
吗啉代
‑
碳鎓六氟磷酸盐(comu)(0.321g,7.5
×
10
‑4mol,sigma
‑
aldrich公司制造),使其在室温下氮气气氛下反应1小时。反应结束后,用氯仿(600g)进行稀释,添加饱和碳酸氢钠水溶液(240g),在室温下搅拌15分钟进行清洗。分离水层和有机层后,再次向有机层中添加饱和碳酸氢钠水溶液(240g),在室温下搅拌15分钟进行清洗,回收有机层。向获得的有机层(氯仿溶液)中添加硫酸镁(2.4g),搅拌30分钟进行脱水,然后使用在5a滤纸上铺有oplite的桐山漏斗进行抽滤。在40℃下浓缩获得的滤液,向浓缩物中添加乙酸乙酯(240g)搅拌至均匀,然后加入己烷(120g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后再次溶解于乙酸乙酯(240g)中,加入己烷(120g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用己烷(120g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p1)(me
‑
200gf
‑
fmoc)。产量为5.1g。
[0132]1h
‑
nmr(d6‑
dmso):1.62ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),2.80ppm(m,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.04ppm(m,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.10ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.24ppm(s,3h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.48ppm(m,约1,900h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.20ppm(m,4h),7.33ppm(m,9h),7.66ppm(m,4h,ar),7.88ppm(d,2h,ar),8.27ppm(t,1h)
[0133]
[实施例1
‑
2]
[0134]
[化28]
[0135]
向实施例1
‑
1中所得的me
‑
200gf
‑
fmoc(4.9g,2.3
×
10
‑4mol)中添加n,n
’‑
二甲基甲酰胺(29.4g),在30℃下加温溶解。添加哌啶(1.55g,1.8
×
10
‑2mol,和光纯药工业株式会社制造),在室温下氮气气氛下反应2小时。反应结束后,加入乙酸乙酯(300g)搅拌至均匀,添加己烷(150g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后再次溶解于乙酸乙酯(300g)中,加入己烷(150g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用己烷(150g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p2)(me
‑
200gf
‑
nh2)。产量为3.9g。
[0136]1h
‑
nmr(d6‑
dmso):1.62ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),1.64ppm(宽,1h),2.59ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),2.98ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.10ppm(q,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.24ppm(s,3h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.48ppm(m,约1,900h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),7.24ppm(m,6h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5,
‑
nh
‑
),7.73ppm(t,1h),8.12ppm(宽,1h)
[0137]
[实施例1
‑
3]
[0138]
[化29]
[0139]
向n末端被fmoc基保护的l
‑
谷氨酸(fmoc
‑
glu
‑
oh)(16.0mg,4.3
×
10
‑5mol,渡边化学工业株式会社制造)和实施例1
‑
2中所得的me
‑
200gf
‑
nh2(2.0g,1.0
×
10
‑4mol)中添加脱水n,n
’‑
二甲基甲酰胺(10g),在30℃下加温溶解。然后,添加二异丙基乙胺(19.2μl,1.1
×
10
‑4mol,关东化学株式会社制造)和4
‑
(4,6
‑
二甲氧基
‑
1,3,5
‑
三嗪
‑2‑
基)
‑4‑
甲基吗啉盐酸盐n水合物(dmt
‑
mm)(39.0mg,1.1
×
10
‑4mol,和光纯药工业株式会社制造),在室温下氮气气氛下反应1小时。然后,添加哌啶(0.5g,5.9
×
10
‑3mol,和光纯药工业株式会社制造),在室温下氮气气氛下反应2小时。反应结束后,反应液用甲苯(80g)进行稀释,然后加入己烷(40g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后再次溶解于甲苯(80g)中,加入己烷(40g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回
收析出物,然后用己烷(40g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p3)(nh2‑
e(fg
‑
200me)2)。产量为1.6g。分子量示于表1中。hplc:胺纯度92%。
[0140]1h
‑
nmr(d6‑
dmso):1.54ppm(m,2h,
‑
nh
‑
co
‑
ch(nh2)
‑
ch2‑
ch2‑
),1.62ppm(m,4h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
),1.97ppm(m,2h,
‑
nh
‑
co
‑
ch(nh2)
‑
ch2‑
ch2‑
),2.74ppm(dd,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),2.81ppm(dd,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),3.11ppm(m,11h),3.24ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.49ppm(m,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),4.57ppm(m,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.25ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.74ppm(m,2h),8.44ppm(m,2h),8.61ppm(m,2h)
[0141]
[实施例2]化合物(p4)(ma
‑
e(fg
‑
200me)2)的合成
[0142]
[化30]
[0143]
将实施例1中所得的化合物(p3)(200mg,5.0
×
10
‑6mol)溶解于乙腈(160mg)和甲苯(1.0g)中。然后,添加n
‑
甲基吗啉(10mg,1.0
×
10
‑5mol,关东化学株式会社制造)和3
‑
马来酰亚胺丙酸n
‑
羟基琥珀酰亚胺酯(8.0mg,3.0
×
10
‑5mol,大阪合成有机化学研究所株式会社制造),在室温下氮气气氛下和遮光下反应6小时。反应结束后,反应液用含2,6
‑
二叔丁基对甲酚(bht)(10mg)的乙酸乙酯(50g)进行稀释,然后加入己烷(25g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用含bht(5mg)的己烷(25g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p4)(ma
‑
e(fg
‑
200me)2)。产量为137mg。分子量示于表1中。马来酰亚胺纯度为90%(1h
‑
nmr)。
[0144]1h
‑
nmr(d6‑
dmso):1.62ppm(m,6h),1.99ppm(m,2h,
‑
nh
‑
co
‑
ch(nh2)
‑
ch2‑
ch2‑
),2.34ppm(m,2h,
‑
nh
‑
co
‑
ch2‑
ch2‑
马来酰亚胺),2.75ppm(dd,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),2.82ppm(dd,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),3.11ppm(m,11h),3.24ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.04ppm(m,2h,
‑
nh
‑
co
‑
ch2‑
ch2‑
马来酰亚胺),4.49ppm(m,2h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),6.98ppm(s,2h,
‑
co
‑
ch
‑
ch
‑
co
‑
),7.25ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.69ppm(dt,2h),8.04ppm(d,1h),8.29ppm(dd,2h),8.41ppm(dt,2h)
[0145]
[实施例3]化合物(p8)(al
‑
e(fg
‑
200me)2)的合成
[0146]
[化31]
[0147]
[实施例3
‑
1]化合物(p5)(ho
‑
e(fg
‑
200me)2)的合成
[0148]
[化32]
[0149]
将ε
‑
己内酯(114mg,1.0
×
10
‑3mol,东京化成工业株式会社制造)溶解于1n naoh(0.8ml,8.0
×
10
‑4mol,关东化学株式会社制造)中反应2小时,配制成6
‑
羟基己酸水溶液(0.88m)。此外,将实施例1中所得的化合物(p3)(2.0g,5.0
×
10
‑5mol)溶解于乙腈(8.0g)中。然后,将上述6
‑
羟基己酸水溶液(114μl,1.0
×
10
‑4mol)、二异丙基乙胺(20μl,1.2
×
10
‑4mol,关东化学株式会社制造)和dmt
‑
mm(21mg,6.0
×
10
‑5mol,和光纯药工业株式会社制造)添加到上述(p3)的乙腈溶液中,在室温下氮气气氛下反应1小时。反应结束后,将反应液在40℃下浓缩,向获得的浓缩物中添加氯仿(24g)进行溶解。添加饱和碳酸氢钠水溶液(10g),在室温下搅拌15分钟进行清洗。分离水层和有机层后,再次向有机层中添加饱和饱和碳酸氢钠水溶液(10g),在室温下搅拌15分钟进行清洗,回收有机层。向获得的有机层(氯仿溶液)中添加硫酸镁(1.2g),搅拌30分钟进行脱水,然后使用在5a滤纸上铺有oplite的桐山漏斗进行抽滤。在40℃下浓缩获得的滤液,向浓缩物中添加甲苯(50g)并搅拌至均匀,然后加入己烷(25g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后再次溶解于甲苯(50g)中,加入己烷(25g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用含bht(2mg)的己烷(10g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p5)(ho
‑
e(fg
‑
200me)2)。产量为1.5g。
[0150]1h
‑
nmr(cdcl3):1.37ppm(m,2h,ho
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.55ppm(m,4h,ho
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.77ppm(m,4h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),1.85ppm(m,1h),2.01ppm(m,2h,ho
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),3.01ppm(m,1h),3.24ppm(m,8h),3.38ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.03ppm(m,4h),4.14ppm(m,1h),4.48ppm(m,2h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),6.95ppm(宽,1h),7.00ppm(宽,1h),7.26ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.66ppm(宽,1h),8.29ppm(宽,1h)
[0151]
[实施例3
‑
2]化合物(p6)(sc
‑
e(fg
‑
200me)2)的合成
[0152]
[化33]
[0153]
将实施例3
‑
1中所得的化合物(p5)(500mg,1.3
×
10
‑5mol)溶解于二氯甲烷(3.5g)中。然后,添加n,n
’‑
二琥珀酰亚胺基碳酸酯(51mg,2.0
×
10
‑4mol,东京化成工业株式会社制造)和吡啶(24μl,3.0
×
10
‑4mol,关东化学株式会社制造),在室温下氮气气氛下反应8小时。反应结束后,用5%食盐水清洗反应液并加入硫酸镁(0.1g),在25℃下搅拌30分钟,然后使用在5a滤纸上铺有oplite的桐山漏斗进行抽滤。将获得的滤液浓缩后,向浓缩物中添加甲苯(50g)进行溶解,然后加入己烷(25g)并在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后再次溶解于甲苯(50g)中,加入己烷(25g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用含bht(5mg)的己烷(25g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p6)(sc
‑
e(fg
‑
200me)2)。产量为286mg。活性碳酸酯纯度为92%(1h
‑
nmr)。
[0154]1h
‑
nmr(cdcl3):1.38ppm(m,2h,琥珀酰亚胺
‑
oco
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.59ppm(m,2h,琥珀酰亚胺
‑
oco
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.75ppm(m,6h),1.85ppm(m,1h),2.13ppm(m,2h,琥珀酰亚胺
‑
oco
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),2.83ppm(s,4h,
‑
co
‑
ch2‑
ch2‑
co
‑
),3.01ppm(m,1h),3.19ppm(m,6h),3.38ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.03ppm(m,3h),4.18ppm(m,1h),4.31ppm(t,2h,琥珀酰亚胺
‑
oco
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),4.50ppm(m,2h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),6.98ppm(宽,1h),7.15ppm(宽,1h),7.26ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.81ppm(宽,1h),8.37ppm(宽,1h)
[0155]
[实施例3
‑
3]化合物(p7)(de
‑
e(fg
‑
200me)2)的合成
[0156]
[化34]
[0157]
将实施例3
‑
2中所得的化合物(p6)(250mg,6.3
×
10
‑6mol)溶解于氯仿(2g)中。然
后,添加1
‑
氨基
‑
3,3
‑
二乙氧基丙烷(10μl,6.3
×
10
‑5mol,acros organics公司制造),在室温下氮气气氛下反应3小时。反应结束后,反应液用甲苯(25g)进行稀释,加入己烷(12.5g)并在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用含bht(2.5mg)的己烷(12.5g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p47)(de
‑
e(fg
‑
200me)2)。产量为185mg。
[0158]1h
‑
nmr(cdcl3):1.20ppm(t,6h,(ch3‑
ch2‑
o)2‑
ch
‑
),1.32ppm(m,2h,(ch3‑
ch2‑
o)2‑
ch
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.58ppm(m,2h,(ch3‑
ch2‑
o)2‑
ch
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.76ppm(m,4h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),1.82ppm(m,2h,(ch3‑
ch2‑
o)2‑
ch
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),2.11ppm(m,2h,(ch3‑
ch2‑
o)2‑
ch
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),2.16ppm(m,1h),2.70ppm(m,1h),3.06ppm(m,2h),3.25ppm(m,11h),3.38ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.02ppm(m,8h),4.17ppm(m,1h),4.51ppm(m,2h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),4.55ppm(t,1h,(ch3‑
ch2‑
o)2‑
ch
‑
),5.36ppm(宽,1h),6.47ppm(宽,1h),6.98ppm(宽,2h),7.26ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.81ppm(宽,1h),8.36ppm(宽,1h)
[0159]
[实施例3
‑
4]化合物(p8)(al
‑
e(fg
‑
200me)2)的合成
[0160]
[化35]
[0161]
将实施例3
‑
3中所得的化合物(p7)(150mg,3.8
×
10
‑6mol)溶解于调节ph至1.90的磷酸盐缓冲液(2.25g)中,在室温下氮气气氛下反应3小时。反应后,添加0.1n氢氧化钠水溶液(0.89g),调节ph至6.40后,添加氯化钠(0.56g)进行溶解。向获得的溶液中滴加0.1n氢氧化钠水溶液(0.60g),调节ph至7.06后,添加含bht(0.6mg)的氯仿(3g),在室温下搅拌20分钟,将生成物萃取至有机层。分离有机层和水层,回收有机层,然后向水层中再次添加含bht(0.6mg)的氯仿(3g),在室温下搅拌20分钟,将生成物萃取至有机层。在40℃下浓缩第1次和第2次萃取中所得的合并有机层,将获得的浓缩物稀释于甲苯(30g)中,加入己烷(15g)并在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用含bht(3.0mg)的己烷(15g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p8)(al
‑
e(fg
‑
200me)2)。产量为84mg。分子量示于表1中。醛纯度为92%(1h
‑
nmr)。
[0162]1h
‑
nmr(cdcl3):1.32ppm(m,2h,cho
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.57ppm(m,2h,cho
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.76ppm(m,4h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),1.82ppm(m,1h),2.10ppm(m,2h,cho
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),2.16ppm(m,1h),2.71ppm(m,2h,cho
‑
ch2‑
ch2‑
nh
‑
coo
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),3.02ppm(m,1h),3.26ppm(m,8h),3.38ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.01ppm(m,4h),4.16ppm(m,1h),4.49ppm(m,2h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),5.59ppm(宽,1h),6.36ppm(宽,1h),6.93ppm(宽,2h),7.08ppm(宽,1h),7.26ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.80ppm(宽,1h),8.37ppm(宽,1h),9.79ppm(s,1h,cho
‑
ch2‑
ch2‑
nh
‑
coo
‑
)
[0163]
[实施例4]化合物(p9)(nh2o
‑
e(fg
‑
200me)2)的合成
[0164]
[化36]
[0165]
将实施例3
‑
1中所得的化合物(p5)(300mg,7.5
×
10
‑6mol)在30℃下加温溶解于甲苯(2.4g)中,减压下共沸脱水。然后,将浓缩物溶解于氯仿(2.4g)中,添加n
‑
羟基邻苯二甲酰亚胺(7.3mg,4.5
×
10
‑5mol,和光纯药工业株式会社制造)、三苯基膦(35mg,1.4
×
10
‑4mol,关东化学株式会社制造)和偶氮二羧酸二异丙酯(22μl,1.1
×
10
‑4mol,acros organics公司制造),在室温下氮气气氛下反应4小时。反应结束后,向反应液中添加甲醇(9.1μl)并在25℃下搅拌30分钟,在40℃下浓缩。将浓缩物稀释于甲苯(3.0g)中并共沸,然后将浓缩物溶解于甲苯(1.5g)中,添加乙二胺一水合物(24μl,3.0
×
10
‑4mol,关东化学株式会社制造),在室温下氮气气氛下反应1小时。反应结束后,反应液用甲苯(50g)进行稀释,然后加入己烷(25g)并在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用己烷(20g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p9)(nh2o
‑
e(fg
‑
200me)2)。产量为156mg。分子量示于表1中。hplc:氧化胺纯度91%。
[0166]1h
‑
nmr(cdcl3):1.32ppm(m,2h,h2n
‑
o
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.56ppm(m,4h,h2n
‑
o
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),1.76ppm(m,4h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),1.85ppm(m,1h),2.10ppm(m,2h,h2n
‑
o
‑
ch2‑
ch2‑
ch2‑
ch2‑
ch2‑
co
‑
nh
‑
),2.17ppm(m,1h),3.01ppm(m,1h),3.24ppm(m,8h),3.38ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.03ppm(m,2h),4.17ppm(m,1h),4.49ppm(m,2h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),5.37ppm(宽,2h),6.40ppm(宽,1h),6.95ppm(宽,2h),7.12ppm(宽,1h),7.26ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.74ppm(宽,1h),8.31ppm(宽,1h)
[0167]
[实施例5]化合物(p13)(nh2‑
e{e(fg
‑
100me)2}2)的合成
[0168]
[化37]
[0169]
[实施例5
‑
1]化合物(p10)(me
‑
100gf
‑
fmoc)的合成
[0170]
[化38]
[0171]
按照与实施例1
‑
1相同的制造方法,使用n末端被fmoc基保护的l
‑
苯丙氨酰
‑
甘氨酸(fmoc
‑
phe
‑
gly)(533mg,1.2
×
10
‑3mol,渡边化学工业株式会社制造)和末端具有丙基氨基的甲氧基peg(9.9g,1.0
×
10
‑3mol,数均分子量=9,896,日油株式会社制造的“sunbright mepa
‑
10t”)作为原料,获得上述化合物(p10)(me
‑
100gf
‑
fmoc)。产量为9.2g。
[0172]1h
‑
nmr(d6‑
dmso):1.62ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),2.80ppm(m,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.04ppm(m,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.10ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.24ppm(s,3h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.48ppm(m,约900h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.20ppm(m,4h),7.33ppm(m,9h),7.66ppm(m,4h,ar),7.88ppm(d,2h,ar),8.27ppm(t,1h)
[0173]
[实施例5
‑
2]化合物(p11)(me
‑
100gf
‑
nh2)的合成
[0174]
[化39]
[0175]
按照与实施例1
‑
2相同的制造方法,使用实施例5
‑
1中所得的化合物(p10)(9.2g,4.6
×
10
‑4mol)进行脱保护反应,获得上述化合物(p11)(me
‑
100gf
‑
nh2)。产量为8.7g。
[0176]1h
‑
nmr(d6‑
dmso):1.62ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),1.64ppm(宽,1h),2.59ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),2.98ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.10ppm(q,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.24ppm(s,3h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.48ppm(m,约900h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),7.24ppm(m,6h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5,
‑
nh
‑
),7.73ppm(t,1h),8.12ppm(宽,1h)
[0177]
[实施例5
‑
3]化合物(p12)(nh2‑
e(fg
‑
100me)2)的合成
[0178]
[化40]
[0179]
按照与实施例1
‑
3相同的制造方法,使用n末端被fmoc基保护的l
‑
谷氨酸(fmoc
‑
glu
‑
oh)(135mg,3.7
×
10
‑4mol,渡边化学工业株式会社制造)和实施例5
‑
2中所得的化合物(p11)(8.5g,8.5
×
10
‑4mol)作为原料,连续进行反应和脱保护,获得上述化合物(p12)(nh2‑
e(fg
‑
100me)2)。产量为6.6g。hplc:胺纯度95%。
[0180]1h
‑
nmr(d6‑
dmso):1.54ppm(m,2h,
‑
nh
‑
co
‑
ch(nh2)
‑
ch2‑
ch2‑
),1.62ppm(m,4h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
),1.97ppm(m,2h,
‑
nh
‑
co
‑
ch(nh2)
‑
ch2‑
ch2‑
),2.74ppm(dd,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),2.81ppm(dd,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),3.11ppm(m,11h),3.24ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约1,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.49ppm(m,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),4.57ppm(m,1h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.25ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.74ppm(m,2h),8.44ppm(m,2h),8.61ppm(m,2h)
[0181]
[实施例5
‑
4]化合物(p13)(nh2‑
e{e(fg
‑
100me)2}2)的合成
[0182]
[化41]
[0183]
按照与实施例1
‑
3相同的制造方法,使用n末端被fmoc基保护的l
‑
谷氨酸(fmoc
‑
glu
‑
oh)(15.2mg,4.1
×
10
‑5mol,渡边化学工业株式会社制造)和实施例5
‑
3中所得的化合物(p12)(2.0g,1.0
×
10
‑4mol)作为原料,连续进行反应和脱保护,获得上述化合物(p13)(nh2‑
e{e(fg
‑
100me)2}2)。产量为1.2g。分子量示于表1中。hplc:胺纯度94%。
[0184]1h
‑
nmr(d6‑
dmso):1.62ppm(m,14h),2.00ppm(m,6h,
‑
nh
‑
co
‑
ch(nh2)
‑
ch2‑
ch2‑
),2.78ppm(m,4h),3.11ppm(m,14h),3.24ppm(s,16h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,600h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.19ppm(m,2h),4.51ppm(m,4h),7.25ppm(m,20h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.71ppm(m,4h),7.89ppm(m,1h),8.45ppm(m,9h)
[0185]
[实施例6]化合物(p16)(nh2‑
e(gflg
‑
200me)2)的合成
[0186]
[化42]
[0187]
[实施例6
‑
1]化合物(p14)(me
‑
200glfg
‑
fmoc)的合成
[0188]
[化43]
[0189]
按照与实施例1
‑
1相同的制造方法,使用n末端被fmoc基保护的l
‑
甘氨酰
‑
苯丙氨酰
‑
亮氨酰
‑
甘氨酸(fmoc
‑
gly
‑
phe
‑
leu
‑
gly)(66mg,1.1
×
10
‑4mol,渡边化学工业株式会社制造)和末端具有丙基氨基的甲氧基peg(1.5g,7.1
×
10
‑5mol,数均分子量=21,120,日油株式会社制造的“sunbright mepa
‑
20t”)作为原料,获得上述化合物(p14)(me
‑
200glfg
‑
fmoc)。产量为1.2g。
[0190]1h
‑
nmr(cdcl3):0.89ppm(d,3h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2)、0.91ppm(d,3h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1.53ppm(m,2h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1,70ppm(m,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1.80ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.10ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.18ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.33ppm(m,7h),3.74ppm(m,约1,900h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.31ppm(宽,1h),4.55ppm(t,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),6.91ppm(宽,1h),7.00ppm(宽,1h),7.28ppm(m,5h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),7.33ppm(t,2h,ar),7.41ppm(m,3h,ar),7.73ppm(m,3h,ar),7.89ppm(d,2h,ar),7.98ppm(宽,1h)
[0191]
[实施例6
‑
2]化合物(p15)(me
‑
200glfg
‑
nh2)的合成
[0192]
[化44]
[0193]
按照与实施例1
‑
2相同的制造方法,使用实施例6
‑
1中所得的化合物(p14)(1.2g,5.7
×
10
‑5mol)进行脱保护反应,获得上述化合物(p15)(me
‑
200glfg
‑
nh2)。产量为1.0g。
[0194]1h
‑
nmr(cdcl3):0.89ppm(d,3h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2)、0.91ppm(d,3h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1.53ppm(m,2h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1,70ppm(m,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1.80ppm(m,2h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.10ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.18ppm(dd,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),3.33ppm(m,7h),3.74ppm(m,约1,900h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.31ppm(宽,1h),4.55ppm(t,1h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),6.91ppm(宽,1h),7.00ppm(宽,1h),7.28ppm(m,5h,
‑
nh
‑
co
‑
ch
‑
ch2‑
c6h5),7.98ppm(宽,1h)
[0195]
[实施例6
‑
3]
化合物(p16)(nh2‑
e(gflg
‑
200me)2)的合成
[0196]
[化45]
[0197]
按照与实施例1
‑
3相同的制造方法,将n末端被fmoc基保护的l
‑
谷氨酸(fmoc
‑
glu
‑
oh)(8.3mg,2.3
×
10
‑5mol,渡边化学工业株式会社制造)和实施例6
‑
2中所得的化合物(p15)(1.0g,4.8
×
10
‑5mol)用作原料,连续进行反应和脱保护,获得上述化合物(p16)(nh2‑
e(gflg
‑
200me)2)。产量为0.5g。分子量示于表1中。hplc:胺纯度90%。
[0198]1h
‑
nmr(cdcl3):0.89ppm(d,6h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2)、0.91ppm(d,6h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1.53ppm(m,4h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1,70ppm(m,2h,
‑
nh
‑
co
‑
ch
‑
ch2‑
ch(ch3)2),1.77ppm(m,4h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),1.85ppm(m,1h),3.01ppm(m,1h),3.24ppm(m,8h),3.38ppm(s,6h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.64ppm(m,约3,800h,
‑
co
‑
nh
‑
ch2‑
ch2‑
ch2‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.03ppm(m,4h),4.14ppm(m,1h),4.48ppm(m,2h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),6.95ppm(宽,1h),7.00ppm(宽,1h),7.26ppm(m,10h,
‑
co
‑
nh
‑
ch
‑
ch2‑
c6h5),7.66ppm(宽,2h),8.29ppm(宽,2h)
[0199]
[比较例1]化合物(p18)(ly
‑
400nh2)的合成
[0200]
[化46]
[0201]
[比较例1
‑
1]化合物(p17)(ly
‑
400bo)的合成
[0202]
[化47]
[0203]
将已上市的聚乙二醇修饰剂中使用的赖氨酸骨架的2分支型聚乙二醇活化酯(3.0g,7.5
×
10
‑5mol,数均分子量=39,700,日油株式会社制造的“sunbright ly
‑
400ns”)在40℃下加温溶解于甲苯(15g)中,添加n
‑
(叔丁氧基羰基)
‑
1,2
‑
二氨基乙烷(48μl,3.0
×
10
‑4mol,东京化成工业株式会社制造),在40℃下在氮气气氛下反应1小时。反应结束后,反应液用乙酸乙酯(12g)进行稀释,然后加入己烷(14g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后再次溶解于乙酸乙酯(27g)中,加入己烷(14g),在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用己烷(30g)进行清洗,使用5a滤纸进行抽滤,通过真空干燥获得上述化合物(p17)(ly
‑
400bo)。产量为2.7g。
[0204]1h
‑
nmr(cdcl3):1.37ppm(m,2h),1.43ppm(s,9h,
‑
ch2‑
nh
‑
co2‑
c
‑
(ch3)3),1.51ppm(m,2h),3.15ppm(m,2h),3.38ppm(s,6h,
‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.65ppm(m,约3,650h,
‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.21ppm(m,4h)
[0205]
[比较例1
‑
2]化合物(p18)(ly
‑
400nh2)的合成
[0206]
[化48]
[0207]
将比较例1
‑
1中所得的化合物(p17)(1.0g,2.5
×
10
‑6mol)溶解于离子交换水(4.0g)中,添加甲磺酸(57μl,8.8
×
10
‑4mol,关东化学株式会社制造),在40℃下在氮气气氛下反应6小时。反应后,用离子交换水(6.0g)进行稀释,添加1n氢氧化钠水溶液(0.9g),调节ph至12后,添加氯化钠(2.5g)进行溶解。向获得的溶液中添加含bht(1.0mg)的氯仿(10g),在室温下搅拌20分钟,将生成物萃取至有机层。分离有机层和水层,回收有机层,然后在40℃下进行浓缩,获得的浓缩物用甲苯(30g)进行稀释,加入己烷(15g)并在室温下搅拌15分钟使生成物析出。使用5a滤纸进行抽滤,回收析出物,然后用含bht(3.0mg)的己烷(15g)进行清洗,使用5a滤纸进行抽滤,真空干燥而获得上述化合物(p18)(ly
‑
400nh2)。产量为0.7g。分子量示于表1中。hplc:胺纯度97%。
[0208]1h
‑
nmr(cdcl3):1.37ppm(m,2h),1.51ppm(m,2h),3.15ppm(m,2h),3.38ppm(s,6h,
‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),3.65ppm(m,约3,650h,
‑
o
‑
(ch2‑
ch2‑
o)n
‑
ch3),4.21ppm(m,4h)
[0209]
[表1] 样品名分子量(mn)实施例1化合物(p3)42,417实施例2化合物(p4)42,534实施例3化合物(p8)42,334实施例4化合物(p9)42,190实施例5化合物(p13)38,234实施例6化合物(p16)42,398比较例1化合物(p18)39,654
[0210]
[实施例7]在血清中的稳定性试验向1.5ml的微型离心管中,加入小鼠或人血清1ml,添加各种聚乙二醇衍生物至浓度成为5.0mg/ml。在37℃下孵育96小时后,取样200μl,向其中添加乙腈,用旋涡混合器(vortex)搅拌1分钟,使血清中的蛋白质析出,离心分离后,回收上清液。接着为了去除脂肪酸等疏水性物质,向回收液中添加己烷,用旋涡混合器搅拌1分钟,离心分离后,回收下层。将该溶液在真空条件下进行浓缩,从血清中回收聚乙二醇衍生物。然后,进行gpc分析,计算分解性聚乙二醇衍生物的分解率。分解率按下式进行计算:分解率=(试验前的40kda的峰面积%
‑
试验后的40kda的峰面积%)
÷
(试验前的40kda的峰面积%)
×
100结果示于下表2中。
[0211]
[表2]
[0212]
根据表2,作为分解性聚乙二醇衍生物的化合物(p3)、(p13)、(p16)与作为非分解性的聚乙二醇衍生物的化合物(p18)、甲氧基peg胺40kda同样地,在血清中未观察到分解。即表示该分解性聚乙二醇衍生物在血液中稳定。
[0213]
[实施例8]使用细胞的分解性试验使用培养基rpmi
‑
1640(10%fbs pn/st)10ml,向100mm培养皿中接种10
×
106细胞的raw264.7,在37℃下培养24小时后,更换到溶解有各种聚乙二醇衍生物至浓度成为10mg/ml的培养基,在37℃下培养96小时。培养后,将细胞溶解于1%sds溶液中,用磷酸盐缓冲生
理盐水(pbs)进行稀释,向其中添加乙腈,用旋涡混合器搅拌1分钟,使细胞溶解液中的蛋白质析出,离心分离后,回收上清液。接着为了去除脂肪酸等疏水性物质,向回收液中添加己烷,用旋涡混合器搅拌1分钟,离心分离后,回收下层。将该溶液在真空条件下进行浓缩,从细胞内回收聚乙二醇衍生物。另外,为了确认在用于细胞培养的培养基中的分解,在37℃下仅在溶解有各种聚乙二醇衍生物至浓度成为10mg/ml的培养基中培养96小时,按照与上述相同操作进行聚乙二醇衍生物的回收。然后,进行回收的各种聚乙二醇衍生物的gpc分析,按照与实施例7相同的计算式,计算分解性聚乙二醇衍生物的分解率。结果示于下表3中。此外,化合物(p3)、(p13)的细胞实验前后的gpc图谱分别示于图1和图2、图3和图4中。
[0214]
[表3]
[0215]
根据表3,作为分解性聚乙二醇衍生物的化合物(p3)和(p16)可确认到在细胞内有效地分解(分解率99%),从分子量4万分解至2万。此外,在化合物(p13)中,可确认到以分解率99%从分子量4万分解至1万。这些分解性聚乙二醇衍生物由于在用于细胞培养的培养基中不分解,因此确认在细胞内被特异性分解。另一方面,在作为非分解性的聚乙二醇衍生物的化合物(p18)和甲氧基peg胺40kda中,均未观察到在细胞内的分解。
[0216]
[实施例9]鲑降钙素(sct)的peg化将氨基酸序列:csnlstcvlg klsqelhklq typrtntgsg tp(序列号:1)的鲑降钙素(sct)(0.5mg,1.5
×
10
‑7mol,株式会社ph japan制造)溶解于100mm乙酸钠缓冲液(ph5.0)中,加入实施例3中所得的化合物(p8)或甲氧基peg醛40kda(18mg,4.5
×
10
‑7mol)和作为还原剂的2
‑
甲基吡啶硼烷(2.0
×
10
‑6mol),调节sct浓度至1.0mg/ml,在4℃下使其反应24小时。然后,使用10mm乙酸钠缓冲液(ph5.0)对反应液进行透析,通过使用hitrap sp hp(5ml,ge healthcare公司制造)的离子交换色谱法进行提纯,获得sct
‑
e(fg
‑
200me)2或甲氧基peg40kda
‑
sct。各自的摩尔收率为36%、49%。
[0217]
rplc分析装置:waters公司制造的“alliance”检测器:uv(280nm)色谱柱:inertsil wp300 c18(gl sciences公司)流动相a:0.05%tfa
‑
h2o
流动相b:0.05%tfa
‑
acn梯度:按照b30%(0min)、b40%(5min)、b50%(15min)、b100%(16min)、b100%(20min)的顺序进行变更流速:1.0ml/min色谱柱温度:40℃在上述rplc分析条件下,计算peg化sct的纯度。结果示于图5中。sct
‑
e(fg
‑
200me)2的rplc纯度:99%甲氧基peg40kda
‑
sct的rplc纯度:99%
[0218]
maldi
‑
tof
‑
ms分析装置:bruker公司制造的“autoflex3”样品:0.5mg/ml,pbs溶液基质:α
‑
氰基
‑4‑
羟基肉桂酸(chca)饱和溶液(0.01%tfa
‑
h2o:acn=2:1)混合样品(1μl)和基质(19μl),取1μl在靶板上点样。在上述maldi
‑
tof
‑
ms分析条件下,测定原料的peg和peg化sct的分子量。图6中,合并示出作为原料的化合物(p8)和sct
‑
e(fg
‑
200me)2的maldi
‑
tof
‑
ms的结果。sct
‑
e(fg
‑
200me)2的分子量:46,405化合物(p8)的分子量:43,136图7中,合并示出作为原料的甲氧基peg醛40kda和甲氧基peg40kda
‑
sct的maldi
‑
tof
‑
ms的结果。甲氧基peg40kda
‑
sct的分子量46,427甲氧基peg醛40kda的分子量43,303根据图7,可确认到peg化sct的分子量相比原料的peg衍生物的分子量,大致仅sct的分子量的部分增加。
[0219]
sds
‑
page分析试剂盒:赛默飞世尔科技公司制造的nupage(注册商标)bis
‑
tris precast gel(凝胶浓度4
‑
12%)染色液:考马斯亮蓝溶液(cbb溶液)或碘染色溶液(bacl2 i2溶液)按照上述sds
‑
page试剂盒的推荐测定条件,进行peg化sct的评价。结果示于图8中。根据图8,在peg化sct中,在使蛋白质和肽选择性染色的cbb染色下,可观察到条带,进一步地,在使聚乙二醇染色的碘染色中,也可观察到条带。在两种染色下观察到条带,可确认到聚乙二醇衍生物与sct键合。
[0220]
[实施例10]人生长激素(hgh)的peg化将氨基酸序列:mfptiplsrl fdnamlrahr lhqlafdtyq efeeayipke qkysflqnpq tslcfsesip tpsnreetqq ksnlellris llliqswlep vqflrsvfan slvygasdsn vydllkdlee giqtlmgrle dgsprtgqif kqtyskfdtn shnddallkn ygllycfrkd mdkvetflri vqcrsvegsc gf(序列号:2)的人生长激素(hgh)(0.4mg,1.8
×
10
‑8mol,shenandoah biotechnology公司制造)溶解于
100mm乙酸钠缓冲液(ph5.5)中,加入实施例3中所得的化合物(p8)或甲氧基peg醛40kda(3.6mg,9.0
×
10
‑8mol)和作为还原剂的氰基硼氢化钠(9.0
×
10
‑7mol),调节hgh浓度至1.0mg/ml,在25下使其反应24小时。然后,使用10mm乙酸钠缓冲液(ph4.7)对反应液进行透析,通过使用hitrap sp hp(5ml,ge healthcare公司制造)的离子交换色谱法进行提纯,获得hgh
‑
e(fg
‑
200me)2或甲氧基peg40kda
‑
hgh。各自的摩尔收率为28%、32%。
[0221]
rplc分析装置:waters公司制造的“alliance”检测器:uv(280nm)色谱柱:inertsil wp300 c18(gl sciences公司)流动相a:0.1%tfa
‑
h2o流动相b:0.1%tfa
‑
acn梯度:按照b40%(0min)、b80%(25min)、b90%(27min)、b40%(27.1min)的顺序进行变更流速:1.0ml/min色谱柱温度:25℃在上述rplc分析条件下,peg化hgh的纯度计算。结果示于图9中。hgh
‑
e(fg
‑
200me)2的rplc纯度:90%甲氧基peg40kda
‑
hgh的rplc纯度:97%
[0222]
maldi
‑
tof
‑
ms分析装置:bruker公司制造的“autoflex3”样品:0.5mg/ml,pbs溶液基质:肉桂酸(sa)饱和溶液(0.01%tfa
‑
h2o:acn=2:1)混合样品(1μl)和基质(19μl),取1μl在靶板上点样在上述maldi
‑
tof
‑
ms分析条件下,测定peg化hgh的分子量。结果示于图10和图11中。hgh
‑
e(fg
‑
200me)2的分子量:65,584甲氧基peg40kda
‑
hgh的分子量:65,263根据图10和图11,可确认到peg化hgh的分子量相比原料的peg衍生物的分子量(参考图6和图7的下图),大致仅hgh的分子量的部分增加。
[0223]
sds
‑
page分析试剂盒:赛默飞世尔科技公司制造的nupage(注册商标)bis
‑
tris precast gel(凝胶浓度4
‑
12%)染色液:考马斯亮蓝溶液(cbb溶液)或碘染色溶液(bacl2 i2溶液)按照上述sds
‑
page试剂盒的推荐测定条件,进行peg化hgh的评价。结果示于图12中。根据图12,在peg化hgh中,在使蛋白质和肽选择性染色的cbb染色下,可观察到条带,进一步地,在使聚乙二醇染色的碘染色中,也可观察到条带。此外,在两种染色下观察到条带,可确认到聚乙二醇衍生物与hgh键合。
[0224]
[实施例11]粒细胞集落刺激因子(gcsf)的peg化
将氨基酸序列:tplgpasslp qsfllkcleq vrkiqgdgaa lqeklcatyk lchpeelvll ghslgipwap lsscpsqalq lagclsqlhs glflyqgllq alegispelg ptldtlqldv adfattiwqq meelgmapal qptqgampaf asafqrragg vlvashlqsf levsyrvlrh laqp(序列号:3)的粒细胞集落刺激因子(gcsf)(0.1mg,5.3
×
10
‑9mol,peprotech公司制造)溶解于10mm乙酸钠缓冲液(ph4.6,含5%山梨糖醇)中,加入实施例3中所得的化合物(p8)和作为还原剂的氰基硼氢化钠(5.3
×
10
‑7mol),调节gcsf浓度至2.0mg/ml,在4℃下使其反应24小时。然后,使用10mm乙酸钠缓冲液(ph4.6)对反应液进行稀释,通过使用hitrap sp hp(5ml,ge healthcare公司制造)的离子交换色谱法进行提纯,获得gcsf
‑
e(fg
‑
200me)2。摩尔收率41%。
[0225]
rplc分析装置:waters公司制造的“alliance”检测器:uv(280nm)色谱柱:inertsil wp300 c18(gl sciences公司)流动相a:0.1%tfa
‑
h2o流动相b:0.1%tfa
‑
acn梯度:按照b40%(0min)、b70%(25min)、b90%(27min)、b40%(29min)的顺序进行变更流速:1.0ml/min色谱柱温度:40℃在上述rplc分析条件下,计算peg化gcsf的纯度。gcsf
‑
e(fg
‑
200me)2的rplc纯度:97%
[0226]
maldi
‑
tof
‑
ms分析装置:bruker公司制造的“autoflex3”样品:0.5mg/ml,pbs溶液基质:肉桂酸(sa)饱和溶液(0.01%tfa
‑
h2o:acn=2:1)混合样品(1μl)和基质(19μl),取1μl在靶板上点样在上述maldi
‑
tof
‑
ms分析条件下,测定peg化gcsf的分子量。gcsf
‑
e(fg
‑
200me)2的分子量:62,199根据上述maldi
‑
tof
‑
ms分析结果,可确认到peg化gcsf的分子量相比原料的peg衍生物的分子量,大致仅gcsf的分子量的部分增加。
[0227]
sds
‑
page分析试剂盒:赛默飞世尔科技公司制造的nupage(注册商标)bis
‑
tris precast gel(凝胶浓度4
‑
12%)染色液:考马斯亮蓝溶液(cbb溶液)或碘染色溶液(bacl2 i2溶液)按照上述sds
‑
page试剂盒的推荐测定条件,进行peg化gcsf的评价。根据上述sds
‑
page分析结果,在peg化gcsf中,在使蛋白质和肽选择性染色的cbb染色下,可观察到条带,进一步地,在使聚乙二醇染色的碘染色中,也可观察到条带。此外,在两种染色下观察到条带,确认到聚乙二醇衍生物与gcsf键合。
[0228]
[实施例12]
peg化鲑降钙素(sct)的生理活性评价在实施例9中所得的作为键合有分子量4万的分解性聚乙二醇衍生物的sct的sct
‑
e(fg
‑
200me)2和具有键合有非分解性的甲氧基peg40kda的甲氧基peg40kda
‑
sct、未修饰的sct和pbs这4群组中,用动物实验对它们的生理活性进行了对比评价。小鼠品种为balb/c(8周龄,雄性),peg化sct溶液和未修饰的sct溶液使用pbs配制为以sct浓度计成为8.0μg/ml,以sct的给药量计成为40μg/kg进行给药。在1、6、24小时时从小鼠采血收集血浆,使用钙e
‑
test wako(富士胶卷和光纯药工业株式会社制造)测定钙浓度。其结果示于图16中。根据图16,相比于pbs的群组,全部的sct均使钙浓度显著降低。未修饰的sct从给药6小时后可观察到钙浓度的上升,但sct
‑
e(fg
‑
200me)2和甲氧基peg40kda
‑
sct可知继续保持较低的钙浓度。确认到因peg化使sct的血液半衰期变长,生理活性未受影响而得以保持。
[0229]
[实施例13]利用动物实验的空泡形成评价试验使用作为在末端具有氨基的分子量4万的分解性聚乙二醇衍生物的化合物(p3)nh2‑
e(fg
‑
200me)2和具有非分解性的甲氧基peg胺40kda,进行利用动物实验的空砲形成评价。小鼠品种为balb/c(8周龄,雄性),聚乙二醇溶液使用生理盐水配制聚乙二醇衍生物至浓度成为100mg/ml,经由小鼠尾静脉给药20μl。持续进行每周3次、4周的连续给药,给药结束后,用4%多聚甲醛水溶液对小鼠进行灌注固定,制作石蜡切片。进行he染色和利用抗peg抗体的免疫染色,评价脑脉络丛上皮细胞中的空泡形成。作为免疫染色,使用免疫染色试剂盒(bond refine polymer detection kit,徕卡公司制造)和抗peg抗体(b
‑
47抗体,abcam公司制造)来实施。将进行利用抗peg抗体的免疫染色的脑脉络丛切片的图像示于图13(甲氧基peg胺40kda)和图14(nh2‑
e(fg
‑
200me)2)中。其结果,作为分解性聚乙二醇的nh2‑
e(fg
‑
200me)2相比于甲氧基peg胺40kda,显著地抑制了空泡的形成。此外,本实施例中给药的聚乙二醇的量终归是为了评价空泡化而优化后的量,相比于该技术领域中的通常的聚乙二醇的给药量,是非常多的量。
[0230]
[实施例14]利用动物实验的聚乙二醇的蓄积性评价试验使用在作为末端具有氨基的分子量4万的分解性聚乙二醇衍生物的化合物(p3)nh2‑
e(fg
‑
200me)2和具有非分解性的甲氧基peg胺20kda、甲氧基peg胺40kda和作为对照的pbs,进行利用动物实验的聚乙二醇的蓄积性评价。小鼠品种为balb/c(8周龄,雄性),聚乙二醇溶液使用生理盐水配制聚乙二醇衍生物至浓度成为62.5mg/ml,经由小鼠尾静脉给药100μl。持续进行每周3次、4周的连续给药,给药结束后,用4%多聚甲醛水溶液对小鼠进行灌注固定,制作石蜡切片。进行利用抗peg抗体的免疫染色,评价脑脉络丛上皮细胞中的蓄积性。将进行免疫染色后的各脑脉络丛切片的图像示于图15中。根据图15,在给予不含聚乙二醇的pbs的小鼠脉络丛切片中未被染色,与此相对,在作为非分解性的甲氧基peg胺40kda中,确认到切片大范围被染色为褐色。该染色部分表示peg出现蓄积。另一方面,在作为分解性聚乙二醇的nh2‑
e(fg
‑
200me)2的切片中,被染成褐色的部分较少,显示出与分子量为其一半的甲氧基peg胺20kda相同的蓄积。作为结果,分解
性聚乙二醇由于其分解性,相比于作为相同分子量的非分解性的甲氧基peg胺40kda,显著地抑制了聚乙二醇在组织中的蓄积。此外,本实施例中给药的聚乙二醇的量终归是为了评价蓄积性而优化后的量,相比于该技术领域中的通常的聚乙二醇的给药量,是非常多的量。产业上的可利用性
[0231]
本发明的分解性聚乙二醇衍生物是不引起细胞的空泡的高分子量的聚乙二醇衍生物,能够有效地用于对生物相关物质进行修饰的用途、在生物体内血液中稳定且在细胞内被分解。本技术以在日本技术的日本专利特愿2019
‑
069450(申请日:2019年3月29日)为基础,其内容全部包含在本说明书中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。