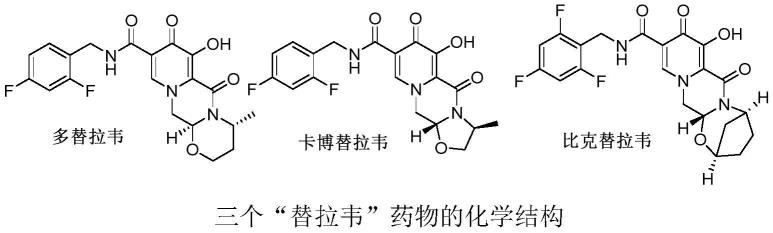
1.本发明属于化学原料药的制备合成领域。具体涉及到连续一锅法制备三个“替拉韦”抗艾滋病药物(包括卡博替拉韦、多替拉韦和比克替拉韦)的合成工艺。
背景技术:
2.hiv病毒是一种逆转录病毒,与其增殖有关的酶有逆转录酶,蛋白酶及整合酶。20世纪80到90年代hiv病毒逆转录酶抑制剂及蛋白酶抑制剂的出现,使艾滋病从一种迅速导致死亡的疾病转变为一种慢性疾病,但是这两类药物存在一些缺陷,例如药物毒副作用、耐药性。整合酶抑制剂是最新一代抗艾滋病药物。dolutegravir(本文英译为多替拉韦)由viivhealthcare公司于2013年首次获得美国fda批准上市,bictegravir(本文英译为比克替拉韦) 由gilead公司开发;cabotegravir(本文英译为卡博替拉韦)也由viiv healthcare公司开发。
[0003][0004]
葛兰素公司在专利申请wo2011119566中报道了一种由4-甲氧基乙酰乙酸甲酯作为起始原料合成多替拉韦的方法,包括以下8步反应,详见下式。
[0005][0006][0007]
上述反应式所示的多替拉韦的合成方法包括了8步反应,反应步骤多,生产成本
高。
[0008]
为了减少药物合成步骤,减低生成成本,本课题组开发了一种连续一锅法制备多替拉韦的合成工艺。由于卡博替拉韦、多替拉韦和比克替拉韦这3个药物在化学结构上极为相似,而且拥有共同的合成中间体-化合物4,该方法被成功应用该三个“替拉韦”抗艾滋病药物的合成制备中。
技术实现要素:
[0009]
本发明提供了一种一锅法制备三个“替拉韦”抗艾滋病药物(包括卡博替拉韦、多替拉韦和比克替拉韦)的合成工艺,包括以下在一锅反应容器中的操作工艺:
[0010]
在第一锅反应容器中制备得到化合物4。
[0011]
在第二锅反应容器中制备得到中间体vi。
[0012]
在第三锅反应容器中制备得到目标“替拉韦”药物viii。
[0013]
进一步的,本发明提供了一种连续一锅法制备3个“替拉韦”药物的合成工艺,包括:
[0014]
1)、4-甲氧基乙酰乙酸甲酯、dmf-dma、草酸二甲酯以及氨基乙醛缩二甲醇“一锅”反应合成得到化合物4;
[0015]
2)、化合物4、手性氨基醇的盐以及酸催化剂在反应溶剂中“一锅”反应合成得到化合物vi;
[0016]
3)、化合物vi、氟代苄胺在耦合剂和有机碱作用下发生偶联,并在路易斯酸催化下“一锅”反应合成得到本技术所述的“替拉韦”药物。如下所示。
[0017][0018]
上述合成化合物4、vi及viii的“一锅”反应指的是在一锅反应容器进行的反应,“一锅”反应的本质在于无需分离和纯化“一锅”反应得到的中间产物。
[0019]
本发明的实施方案中,在第一锅反应容器中制备得到化合物4的工艺是:将4-甲氧基乙酰乙酸甲酯置于反应容器中,在该反应容器中与dmf-dma、草酸二甲酯以及氨基乙醛缩二甲醇“一锅”合成得到化合物4。在合成化合物4的工艺中,无需进行任何中间产物的分离纯化,极大简化了生产工艺的流程和成本。
[0020]
本发明的实施方案中,在第二锅反应容器中制备得到化合物vi的工艺是:将化合物6 置于反应容器中,与相应的手性氨基醇以及酸催化剂在反应溶剂中“一锅”反应合成得到化合物vi。在合成化合物vi的工艺中,无需进行任何中间产物的分离纯化,极大简化了生产工艺的流程和成本。
[0021]
在第二锅反应中,当手性氨基醇分别选自(s)-2-氨基丙醇、(r)-3-氨基丁醇和(1r,3s)-3
‑ꢀ
氨基环戊醇,分别得到用于合成卡博替拉韦、多替拉韦、比克替拉韦的中间体-化合物via,vib, vic。
[0022]
进一步,在第二锅反应中,所述手性氨基醇的盐选自盐酸盐,硫酸盐的一种;优选盐酸盐。
[0023]
进一步,在第二锅反应中,所述的酸催化剂选自对甲苯磺酸,甲磺酸,三氟甲磺酸,三氟乙酸中的一种与乙酸形成的混酸,优选甲磺酸与乙酸的混酸。
[0024]
本发明的实施方案中,在第三锅反应容器中制备得到“替拉韦”药物(在本技术中化合物编号为viii)的工艺是:将化合物vi置于反应容器中,与氟代苄胺在耦合剂及有机碱催化剂作用下发生偶联,并在路易斯酸(例如mgbr2、licl或libr)作用下“一锅”反应合成得到本技术所述的“替拉韦”药物viii。在合成化合物vi的工艺中,无需进行任何中间产物的分离纯化,极大简化了生产工艺的流程和成本。
[0025]
在第二锅反应中,当氟代苄胺选自2,4-二氟苯胺,得到用于合成卡博替拉韦(化合物 viiia)及多替拉韦(化合物viiib),当氟代苄胺选自2,4,6-三氟苯胺,得到用于合成比克替拉韦(化合物viiic),详见以下反应式。
[0026]
进一步,在第三锅反应中,所述耦合剂选自1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐 (edci)、二环己基碳二亚胺(dcc)、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)、1-羟基苯并三唑(hobt)、n,n'-羰基二咪唑(cdi)中的一种,优选edci。
[0027]
进一步,在第三锅反应中,所述有机碱催化剂选自三乙烯二胺(dabco),1,8-二氮杂二环十一碳-7-烯(dbu),4-二甲氨基吡啶(缩写为dmap),吡啶;优选dmap。
[0028]
进一步,在第三锅反应中,所述路易斯酸选自mgbr2、licl、libr中的一种,优选mgbr2, libr。
[0029]
在上述合成及制备工艺中,反应溶剂、反应温度、反应时间本领域技术人员可根据有机化学的基本原理进行适当选择。例如,反应溶剂可以依据反应对温度、溶剂极性的需求,从 n,n-二甲基甲酰胺(缩写dmf)、二甲基亚砜(缩写dmso)、二氯甲烷(缩写dcm)、氯仿、乙腈、四氢呋喃中等选取。反应温度可依据反应类型适当选取。反应中,可以用色谱法、液质连用谱来监控反应进程。色谱法中,可适用薄层色谱,还可用气相色谱法或液相色谱法如 hplc代替等。反应时间可通过薄层层析tlc、高效液相色谱法hplc或lc-ms液相质谱联用等监控手段追踪反应情况得出。本发明中,(s)-2-氨基丙醇、(r)-3-氨基丁醇和(1r,3s)-3
‑ꢀ
氨基环戊醇也可以选用其盐酸盐。
[0030]
本发明的有益之处在于:本发明提供了式i所示的3个“替拉韦”药物新的合成工艺中,在上述的合成化合物4、vi及viii的一锅法工艺中,每一锅反应均无需进行任何中间产物的分离纯化,极大简化了生产工艺的流程和成本,易于工业化生产等优点。
具体实施方式
[0031]
以下将通过具体实施例进一步阐述本发明,但并不用于限制本发明的保护范围。在不脱离本发明构思的前提下,本领域技术人员可对权利要求的各参数或条件做出的改进或组合,这些改进或组合也应视为本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
[0032]
本发明中使用的4-甲氧基乙酰乙酸乙酯,草酸二甲酯、甲磺酸、dmf-dma(n,n-二甲基甲酰胺二甲基缩醛)、(s)-2-氨基丙醇、(r)-3-氨基丁醇和(1r,3s)-3-氨基环戊醇盐酸盐,醋酸、mgbr2,libr来自中国国药集团。
[0033]
以下实施例中,英语缩写h代表小时,tlc代表薄层层析,ml代表毫升,n代表mol/l 的浓度。
[0034]
第一部分:第一锅反应
[0035][0036]
实施例1、化合物4的制备
[0037]
将4-甲氧基乙酰乙酸甲酯8ml(61.80mmol)、n,n-二甲基甲酰胺二甲基缩醛(dmf-dma)9.6ml(72.26mmol)于150ml的圆底烧瓶中室温搅拌1.5小时,溶液由黄色变成棕黄色,tlc薄层层析法跟踪,化合物1(结构式见式1,在本锅反应中无需分离) 的rf=0.137(展开剂:乙酸乙酯:石油醚=5:1,在紫外灯254nm处观察)。在反应液中加入20ml甲醇稀释并加入氨基乙醛缩二甲醇6.68ml(61.31mmol),室温搅拌1h,溶液变为酒红色,tlc薄层层析法跟踪,化合物2(结构式见式1,在本锅反应中无需分离)的rf=0.557 (在紫外灯254nm处观察,展开剂:乙酸乙酯)。停止反应并蒸发浓缩成红棕色油状液体。该残留物加入甲醇45.2ml溶解,草酸二甲酯18.34g(155.34mmol),待草酸二甲酯完全溶解后,控制温度在25℃以下,分批加入氢化锂lih 108.67mmol,溶液成棕红色悬浮液,加入完毕之后,将反应液放置在40℃的油浴锅中反应14小时后,溶液由悬浮液变成砖红色溶液;将所得反应液冷却至-5℃并加入无水氢氧化锂5.94g(123.8mmol)同时控制温度在在 3-5℃的环境中,反应液由砖红色变成橙黄色悬浮液,反应2小时后,加入2n盐酸146.8ml 进行淬灭反应,控制反应温度低于5℃下进行。加入180ml乙酸乙酯进行萃取并将温度升至20℃,抽滤,固体弃去;收集液相并将其分液,收集有机相,同时在有机相中加入90ml 的水,减压浓缩,抽滤,收集滤饼并于50℃真空干燥,得到化合物4为白色固体,rf=0.35 (展开剂:乙酸乙酯,在紫外灯254nm下观察),12.51g,产率:65%,m.p.111.3℃.1h nmr (400mhz,cdcl3)δ=8.40-8.42(m,1h),4.49-4.53(m,1h),4.10-4.14(m,2h),3.98(s,3h),3.97 (s,3h),3.38(s,3h),3.37(s,3h);
13
c nmr(100mhz,cdcl3)δ=174.86,165.99,161.60, 148.65,145.47,136.59,116.53,102.31,60.97,57.26,55.97,53.77.
[0038]
第二部分:第二锅反应
[0039]
实施例2、化合物via(合成cabotegravir的中间体)的制备
[0040]
称取实施例1中得到的中间体缩醛(化合物4,1g,3.2mmol)溶解在50ml ch3cn中。在室温下加入hoac(1ml)和ch3so3h(0.3ml,5.7mmol)的ch3cn(50ml)溶液和(s)-2-氨基丙醇(705mg,9.4mmol),将混合物回流30小时。浓缩混合物,将残余物重新溶解在 ch2cl2(100ml)中。添加1n的hcl(50ml)后,所得混合物分层,水层用ch2cl2(150ml
×ꢀ
2)萃取,合并有机层并浓缩。加入meoh(80ml)并再次浓缩所得混合物。加入meoh(50ml) 后将所得混合物加热回流2小时,逐渐冷却至20℃并在20℃保持15小时。过滤收集产物并真空干燥,得到标题化合物via(746mg,80%),为白色固体,rf=0.69(展开剂:乙酸乙酯,在紫外灯254nm下观察)。1h nmr(400mhz,chloroform-d)δ=8.43(s,1h),5.39(dd,j=9.9, 3.4hz,1h),4.56(dd,j=12.4,3.4hz,1h),4.47
–
4.33(m,2h),4.07(s,3h),3.96(dd,j=12.1, 10.2hz,1h),3.71(t,j=7.5hz,1h),1.42(d,j=6.0hz,3h).
13
c nmr(101mhz,cdcl3)δ=176.16,165.78,153.85,152.48,143.04,131.51,116.04,82.38,73.15,61.60,55.74,52.20,17.02.
[0041]
实施例3、化合物vib(合成dolutegravir的中间体)的制备
[0042]
称取实施例1中得到的中间体缩醛(化合物4,20g,63mmol)溶解在100ml ch3cn中。在室温下加入hoac(100ml)和ch3so3h(1.2ml,18.5mmol)和(r)-3-氨基-1-丁醇 (16.73g,188mmol)的ch3cn溶液(100ml),并将混合物回流15小时。浓缩混合物,并将残余物重新溶解在ch2cl2(200ml)中。添加稀hcl(1n,100ml)并分离各层。水层用 ch2cl2(100ml
×
2)萃取,合并有机层并浓缩。加入meoh(50ml)并将所得混合物加热回流 2小时,有黄色固体从反应液中析出。停止反应,冷却至室温,过滤收集产物并真空干燥,得到标题化合物vib(12.6g,收率65%),为黄色固体。1h nmr(400mhz,cdcl3)δ=15.02(d, j=16.7hz,1h),8.42(d,j=11.8hz,1h),5.29(dd,j=5.4,3.9hz,1h),4.97(q,j=6.4hz,1h), 4.52(t,j=4.5hz,1h),4.43(dd,j=13.6,3.5hz,1h),4.26(dd,j=13.6,5.8hz,1h),4.13(d,j =4.5hz,1h),4.05(d,j=13.0hz,2h),3.98(d,j=2.2hz,3h),3.96(d,j=2.9hz,1h),3.39(s, 3h),2.22-2.09(m,1h),1.52(dd,j=13.9,1.6hz,1h),1.35(d,j=7.0hz,2h);
13
c nmr(100 mhz,cdcl3)δ=173.23,164.17,162.24,149.45,144.64,135.03,130.77,130.73,130.67,119.35, 111.34,111.30,111.13,111.09,103.81,102.73,60.78,56.81,55.72,53.47,36.60,36.56.hrms (esi-tof):309.1166[m 1].
[0043]
实施例4、化合物vic(合成bictegravir的中间体)的制备
[0044]
称取实施例1中得到的中间体缩醛(化合物4,1g,3.2mmol)溶解在50ml ch3cn中。在室温下加入hoac(1ml)和ch3so3h(0.3ml,5.7mmol)的ch3cn(50ml)溶液和(1r,3s)-3
‑ꢀ
氨基环戊醇(1.29g,9.4mmol),将混合物回流30小时。浓缩混合物,将残余物重新溶解在ch2cl2(100ml)中。添加1n的hcl(50ml)后,所得混合物分层,水层用ch2cl2(150ml
×ꢀ
2)萃取,合并有机层并浓缩。加入meoh(80ml)并再次浓缩所得混合物。加入meoh(50ml) 后将所得混合物加热回流2小时,逐渐冷却至20℃并在20℃保持15小时。过滤收集产物并真空干燥,得到标题化合物via(634mg,62%),为微黄固体,rf=0.38(展开剂:dcm:ch3oh =97:3,在紫外灯254nm下观察)。1h nmr(400mhz,dmso-d6)δ=8.70(s,1h),5.40(dd,j =9.7,3.8hz,1h),5.07(d,j=4.0hz,1h),4.70(dd,j=12.9,3.8hz,1h),4.07(dd,j=12.8,9.7 hz,1h),3.81(s,3h),1.89(s,2h),1.88(s,1h),1.76(d,j=10.7hz,2h),1.52(dt,j=12.3,3.3 hz,1h).
13
c nmr(101mhz,dmso-d6)δ=176.04,165.67,153.30,151.69,144.06,133.00, 115.10,76.61,74.13,60.94,54.09,51.01,38.10,28.90,27.86.
[0045]
第三部分:第三锅反应
[0046]
实施例5、化合物viiia(cabotegravir)的制备
[0047]
称取实施例2中得到的化合物via(2g,6.8mmol)溶解在100ml ch3cn中,添加edci (3.87g,20.2mmol)、dmap(334mg,2.7mmol)和2,4-二氟苄胺(1.17g,8.2mmol)以形成悬浮溶液。将混合物在油浴中加热至80℃,2小时后完全溶解。然后加入libr(1g,12mmol)。4 小时后,过滤收集产物,真空干燥,得到白色固体标题化合物viiia(cabotegravir)(2.5g,收率90%)。rf=0.31(dcm:ch3oh=97:3).1h nmr(400mhz,dmso-d6)δ=10.24(s,1h), 7.86(s,1h),7.31(d,j=8.1hz,1h),7.16(t,j=9.4hz,1h),7.02
–
6.92(m,1h),5.16(dd,j= 9.9,4.0hz,1h),4.67(dt,j=20.1,10.3hz,2h),4.29(t,j=7.7hz,1h),4.17(d,j=14.4hz, 2h),3.60(t,j=7.6hz,1h),3.54
–
3.42(m,1h),1.25(d,j=6.0hz,3h).
13
c nmr(101mhz, dmso-d6)δ=177.55,165.46,164.68,159.27,136.60,131.16,115.15,111.83,111.61,109.99, 109.78,104.42,104.16,103.91,81.52,72.67,54.19,51.59,35.90,17.28.
[0048]
实施例6、化合物viiib(dolutegravir的中间体)的制备
[0049]
称取实施例3中得到的化合物vib(10g,32mmol)溶解在300ml ch3cn中,添加edci (20.0g,104mmol)、dmap(1.72g,14.1mmol)和2,4-二氟苄胺(5.85g,40.9mmol)以形成悬浮溶液。将混合物在油浴中加热至80℃,2小时后完全溶解。然后加入libr(5.5g,62mmol)。 4小时后,过滤收集产物,真空干燥,得到白色固体标题化合物viiib(dolutegravir(8.0g,收率59%)。1h nmr(400mhz,cdcl3)δ=12.48(s,1h),10.33(t,j=4hz,1h),8.47(s,1h),7.35 (q,j=4hz,1h),7.20(td,j=8hz,4hz,1h),7.02(dd,j=8hz,4hz,1h),5.42(t,j=8hz,1h), 4.76(t,j=8hz,1h),4.57(d,j=4hz,1h),4.51(d,j=4hz,2h),4.33(q,j=8hz,1h),4.00(t,j =12hz,1h),3.88-3.85(d,j=4hz,1h),2.03-1.94(m,1h),1.51(d,j=12hz,3h).
13
c nmr(100 mhz,cdcl3)δ=174.23,170.89,164.35,164.15,162.22,155.74,153.63,142.92,140.43,130.58, 130.07,121.65,116.88,116.80,115.52,115.42,110.99,110.77,103.51,103.26,102.99,76.43, 76.28,62.36,62.01,60.34,53.07,52.04,48.08,45.05,44.85,36.00,29.12,14.74,14.27.
[0050]
实施例7、化合物viiic(bictegravir)的制备
[0051]
称取实施例4中得到的化合物vic(1.80g,5.6mmol)溶解在100ml ch3cn中,添加edci (3.87g,20.2mmol)、dmap(334mg,2.7mmol)和2,4,6-三氟苄胺(1.32g,,8.2mmol)以形成悬浮溶液。将混合物在油浴中加热至80℃,2小时后完全溶解。然后加入libr(1g,12mmol)。4小时后,过滤收集产物,真空干燥,得到标题化合物viiic(bictegravir)(2.14g,收率85%),为白色固体。rf=0.38(dcm:ch3oh=97:3).1h nmr(400mhz,dmso-d6)δ=12.32(s,1h), 10.25(s,1h),8.25(s,1h),6.61(t,j=8.0hz,2h),5.36(dd,j=9.4,4.0hz,1h),5.25(d,j=9.5 hz,1h),4.65
–
4.58(m,2h),4.24(dd,j=13.1,4.1hz,1h),3.95(dd,j=12.8,9.3hz,1h),3.43 (d,j=1.1hz,1h),2.12
–
1.89(m,6h).13c nmr(101mhz,cdcl3)δ=171.13,163.70,160.45, 155.68,140.21,116.27,115.78,100.48,100.23,100.20,100.18,99.95,99.93,77.28,74.24,53.36, 51.27,37.89,30.59,28.98,27.85.
[0052]
以上所述的仅是本发明的实施例,方案中公知的具体结构及特性等常识在此未作过多描述。应当指出,对于本领域的技术人员来说,在不脱离本发明结构的前提下,还可以作出若干变形和改进,这些也应该视为本发明的保护范围,这些都不会影响本发明实施的
效果和专利的实用性。本技术要求的保护范围应当以其权利要求的内容为准,说明书中的具体实施方式等记载可以用于解释权利要求的内容。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。