1.本发明属于药物化学领域,具体涉及一种治疗偏头痛药物拉司米地坦的制备方法
背景技术:
2.拉司米地坦是一种中枢神经系统渗透性、选择性、5-羟色胺1f(5-ht1f)激动剂口服制剂,且拉司米地坦是地坦(ditan)类药物中第一个用于成人偏头痛急性治疗的药物,其结构上和机制上不同于已获批的偏头痛药物,而且不存在血管收缩活性;如果获批将代表偏头痛治疗的重大创新。2018年11月16日礼来向美国fda提交了拉司米地坦(lasmiditan)新药申请(nda),用于成人患者伴或不伴先兆症状偏头痛的急性治疗。
3.目前报道的合成拉司米地坦的专利中,基本都是要求合成关键中间体式
ⅴ
化合物:
[0004][0005]
已有专利中报道的合成方法或条件苛刻,或采用了危险性较大的锂试剂,工业化生产难度大,且收率不高,导致产品成本过高。
[0006]
因此,本领域需要开发一种产率高、操作简便、易于工业化生产的拉司米地坦制备方法。
技术实现要素:
[0007]
本发明的目的是提供一种产率高、操作简便、易于工业化生产的拉司米地坦制备方法。
[0008]
本发明第一方面,提供了一种式ⅶ化合物的制备方法,所述方法包括步骤:
[0009]
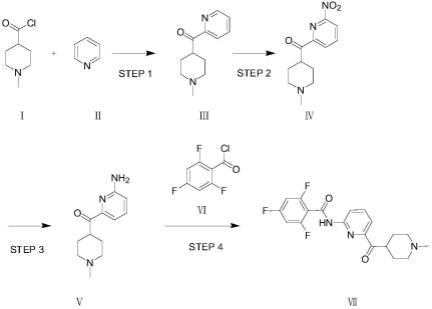
(i)n-甲基-4-哌啶甲酰氯与吡啶发生傅克反应,得到式ⅲ化合物;
[0010][0011]
(ii)式ⅲ与硝酸进行硝基化反应,得到式ⅳ化合物;
[0012][0013]
(iii)式ⅳ化合物进行硝基还原反应,得到式
ⅴ
化合物;和
[0014][0015]
(iv)式
ⅴ
化合物与式ⅵ化合物反应,得到式ⅶ化合物;
[0016][0017]
在另一优选例中,所述方法包括步骤:
[0018]
(i)在惰性溶剂中,在催化剂存在下,n-甲基-4-哌啶甲酰氯与吡啶发生傅克反应,得到式ⅲ化合物;
[0019]
(ii)在浓硝酸和浓硫酸存在下,式ⅲ化合物发生硝基化反应,得到式ⅳ化合物;
[0020]
(iii)在惰性溶剂中,在金属催化剂存在下,h2气氛中,式ⅳ化合物发生硝基还原反应,得到式
ⅴ
化合物;和
[0021]
(iv)在惰性溶剂中,在碱催化剂存在下,式
ⅴ
化合物与式ⅵ化合物反应得到式ⅶ化合物。
[0022]
在另一优选例中,步骤(i)具有一个或多个选自下组的特征:
[0023]
(1)所述反应的催化剂选自下组:无水三氯化铝、无水氯化锌、三氯化铁、四氯化钛,或其组合,较佳的,无水三氯化铝、三氯化铁,或其组合;更佳的,无水三氯化铝;
[0024]
(2)式ⅰ化合物与式ⅱ化合物的摩尔比为1:1~1.5,较佳的,1:1.1~1.3,更佳的,1:1.15-1.25;
[0025]
(3)所述反应的温度为35-50℃,较佳地,40-45℃;和/或
[0026]
(4)所述反应的时间为1-6h,较佳地,1.5-4h。
[0027]
在另一优选例中,所述步骤(i)包括后处理步骤(i-1):
[0028]
(i-1)将步骤(i)的反应物冷却(20
±
5℃)后降温至0
±
4℃,滴加水,控温10-30℃,
搅拌10-30min,静置分层,取有机相,干燥,旋蒸除去溶剂,得到式iii化合物。
[0029]
在另一优选例中,步骤(ii)具有一个或多个选自下组的特征:
[0030]
(1)所述反应为将式iii化合物加入硫酸中,并滴加硝酸和硫酸的混合物;
[0031]
(2)所述式iii化合物与硝酸的摩尔比为1:1-3.0,较佳地,1.5-2.5,更佳地,1.8-2.2;
[0032]
(3)所述反应的温度为-10~5℃,较佳地,-5~0℃;和/或
[0033]
(4)所述反应的时间为0.5-6h,较佳地,1-3h。
[0034]
在另一优选例中,步骤(iii)具有一个或多个选自下组的特征:
[0035]
(1)所述催化剂选自下组:钯炭、铂炭、雷尼镍、铁粉或其组合,较佳的,钯炭;
[0036]
(2)所述惰性溶剂选自下组:c1-c6醇、或c1-c6醇与水的混合溶剂,较佳地,所述混合溶剂中含有80-95%c1-c6醇,较佳地,90-95%醇-水混合溶剂,更佳地,所述醇为甲醇;
[0037]
(3)式iv化合物与催化剂的质量比为1:0.05-0.2,较佳的,1:0.1~0.15;
[0038]
(4)氢气压力为0.1-1mpa,较佳的0.15-0.6mpa,更佳的0.2-0.5mpa;
[0039]
(5)所述反应的温度为15-35℃,较佳地,20~35℃;和/或
[0040]
(6)所述反应的时间为1-8h,较佳地,2-6h。
[0041]
在另一优选例中,所述钯炭的钯含量为3~15wt%,较佳的,5~10wt%,更佳的,8~10wt%。
[0042]
在另一优选例中,步骤(iii)包括步骤:在甲醇和水的混合溶剂中,在8~10wt%的钯炭催化剂存在下,0.2-0.5mpa h2气氛中,15-35℃下,式ⅳ化合物发生硝基还原反应,得到式
ⅴ
化合物。
[0043]
在另一优选例中,步骤(iv)具有一个或多个选自下组的特征:
[0044]
(1)所述碱催化剂选自下组:氨水、三甲胺、三乙胺,碳酸钠,或其组合;
[0045]
(2)所述式
ⅴ
化合物与式ⅵ化合物的摩尔比为1:0.5-2.0,较佳地,0.8-1.5,更佳地,1.0-1.2;
[0046]
(3)所述反应的温度为-5-10℃,较佳地,0~5℃;和/或
[0047]
(4)所述反应的时间为1-8h,较佳地,2-6h。
[0048]
本发明第二方面,提供了一种式v化合物的制备方法,所述方法包括步骤:
[0049]
(i)n-甲基-4-哌啶甲酰氯与吡啶发生傅克反应,得到式ⅲ化合物;
[0050][0051]
(ii)式ⅲ与硝酸进行硝基化反应得到式ⅳ;
[0052][0053]
(iii)式ⅳ化合物进行硝基还原反应得到式
ⅴ
化合物;和
[0054][0055]
在另一优选例中,所述方法包括步骤:
[0056]
(i)在惰性溶剂中,在催化剂存在下,n-甲基-4-哌啶甲酰氯与吡啶发生傅克反应,得到式ⅲ化合物;
[0057]
(ii)在浓硝酸和浓硫酸存在下,式ⅲ化合物发生硝基化反应,得到式ⅳ化合物;
[0058]
(iii)在惰性溶剂中,在金属催化剂存在下,h2气氛中,式ⅳ化合物发生硝基还原反应,得到式
ⅴ
化合物。
[0059]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
具体实施方式
[0060]
本发明人经过广泛而深入的研究,通过大量筛选和测试,提供了一种拉司米地坦的制备方法。具体地,发明人开发了一条非常简单高效的式v化合物的制备方法。在此基础上完成了本发明。
[0061]
术语
[0062]
除非另有定义,否则本文中所用的全部技术术语和科学术语均具有如本发明所属领域普通技术人员通常理解的相同含义。
[0063]
如本文所用,在提到具体列举的数值中使用时,术语“约”意指该值可以从列举的值变动不多于1%。例如,如本文所用,表述“约100”包括99和101和之间的全部值(例如,99.1、99.2、99.3、99.4等)。
[0064]
如本文所用,术语“含有”或“包括(包含)”可以是开放式、半封闭式和封闭式的。换言之,所述术语也包括“基本上由
…
构成”、或“由
…
构成”。
[0065]
如本文所用,术语“室温”、“常温”是指温度为4-40℃,较佳地,25
±
5℃。
[0066]
在本发明的方法中可以使用任何合适的惰性溶剂。代表性的惰性溶剂包括但不限于c1-c6烷基、c1-c6醇、石油醚、环戊烷、苯、甲苯、二甲苯、三氟甲苯,卤代苯如氯苯、氟苯、二氯苯和二氟苯,二氯甲烷、氯仿、dmf、dmso、丙酮、乙酸乙酯、二乙醚、四氢呋喃,或其组合。
在一些实施方案中,溶剂可以是四氢呋喃、氯仿、甲醇、异丙醇,或其组合。
[0067]
式ⅲ化合物的制备
[0068]
本发明提供了一种式ⅲ化合物的制备方法,包括步骤:
[0069]
(i)n-甲基-4-哌啶甲酰氯与吡啶发生傅克反应,得到式ⅲ化合物。
[0070][0071]
具体地,包括(i)在惰性溶剂中,在催化剂存在下,n-甲基-4-哌啶甲酰氯与吡啶发生傅克反应,得到式ⅲ化合物。
[0072]
在另一优选例中,步骤(i)具有一个或多个选自下组的特征:
[0073]
(1)所述反应的催化剂选自下组:无水三氯化铝、无水氯化锌、三氯化铁、四氯化钛,或其组合,较佳的,无水三氯化铝、三氯化铁,或其组合;更佳的,无水三氯化铝;
[0074]
(2)式ⅰ化合物与式ⅱ化合物的摩尔比为1:1~1.5,较佳的,1:1.1~1.3,更佳的,1:1.15-1.25;
[0075]
(3)所述反应的温度为35-50℃,较佳地,40-45℃;和/或
[0076]
(4)所述反应的时间为1-6h,较佳地,1.5-4h。
[0077]
在另一优选例中,所述步骤(i)包括后处理步骤(i-1):
[0078]
(i-1)将步骤(i)的反应物冷却(20
±
5℃)后降温至0
±
4℃,滴加水,控温10-30℃,搅拌10-30min,静置分层,取有机相,干燥,旋蒸除去溶剂,得到式iii化合物。
[0079]
式ⅳ化合物的制备
[0080]
本发明提供了一种式ⅳ化合物的制备方法,包括步骤:
[0081]
(ii)式ⅲ与硝酸进行硝基化反应,得到式ⅳ化合物。
[0082][0083]
具体地,包括(ii)在浓硝酸和浓硫酸存在下,式ⅲ化合物发生硝基化反应,得到式ⅳ化合物。
[0084]
在另一优选例中,步骤(ii)具有一个或多个选自下组的特征:
[0085]
(1)所述反应为将式iii化合物加入硫酸中,并滴加硝酸和硫酸的混合物;
[0086]
(2)所述式iii化合物与硝酸的摩尔比为1:1-3.0,较佳地,1.5-2.5,更佳地,1.8-2.2;
[0087]
(3)所述反应的温度为-10~5℃,较佳地,-5~0℃;和/或
[0088]
(4)所述反应的时间为0.5-6h,较佳地,1-3h。
[0089]
式
ⅴ
化合物的制备
[0090]
本发明提供了一种式
ⅴ
化合物的制备方法,包括步骤:
[0091]
(iii)式ⅳ化合物进行硝基还原反应,得到式
ⅴ
化合物。
[0092][0093]
具体地,包括(iii)在惰性溶剂中,在金属催化剂存在下,h2气氛中,式ⅳ化合物发生硝基还原反应,得到式
ⅴ
化合物。
[0094]
在另一优选例中,步骤(iii)具有一个或多个选自下组的特征:
[0095]
(1)所述催化剂选自下组:钯炭、铂炭、雷尼镍、铁粉或其组合,较佳的,钯炭;
[0096]
(2)所述惰性溶剂选自下组:c1-c6醇、或c1-c6醇与水的混合溶剂,较佳地,所述混合溶剂中含有80-95%c1-c6醇,较佳地,90-95%醇-水混合溶剂,更佳地,所述醇为甲醇;
[0097]
(3)式iv化合物与催化剂的质量比为1:0.05-0.2,较佳的,1:0.1~0.15;
[0098]
(4)氢气压力为0.1-1mpa,较佳的0.15-0.6mpa,更佳的0.2-0.5mpa;
[0099]
(5)所述反应的温度为15-35℃,较佳地,20~35℃;和/或
[0100]
(6)所述反应的时间为1-8h,较佳地,2-6h。
[0101]
在另一优选例中,所述钯炭的钯含量为3~15wt%,较佳的,5~10wt%,更佳的,8~10wt%。
[0102]
特别优选地,步骤(iii)包括步骤:在特定比例的甲醇和水的混合溶剂中(如90-95%甲醇),在8~10wt%的钯炭催化剂存在下,0.2-0.5mpa h2气氛中,15-35℃下,式ⅳ化合物发生硝基还原反应,得到式
ⅴ
化合物。
[0103]
拉司米地坦(式ⅶ化合物)的制备
[0104]
本发明提供了一种式
ⅴ
化合物的制备方法,包括步骤:
[0105]
(iv)式
ⅴ
化合物与式ⅵ化合物反应,得到式ⅶ化合物.
[0106][0107]
具体地,包括(iv)在惰性溶剂中,在碱催化剂存在下,式
ⅴ
化合物与式ⅵ化合物反应得到式ⅶ化合物。
[0108]
在另一优选例中,步骤(iv)具有一个或多个选自下组的特征:
[0109]
(1)所述碱催化剂选自下组:氨水、三甲胺、三乙胺,碳酸钠,或其组合;
[0110]
(2)所述式
ⅴ
化合物与式ⅵ化合物的摩尔比为1:0.5-2.0,较佳地,0.8-1.5,更佳
地,1.0-1.2;
[0111]
(3)所述反应的温度为-5-10℃,较佳地,0~5℃;和/或
[0112]
(4)所述反应的时间为1-8h,较佳地,2-6h。
[0113]
本发明的主要优点包括:
[0114]
本发明提供了一种新的合成路线,避免采用危险性高的锂试剂合成方法,且直接使用了简便易得的原料吡啶直接合成,减小了物料成本,大大增加了产品的附加经济价值。
[0115]
本发明的方法中各步骤反应温度低,原料毒性危险性小,大大提高了反应安全性,与现有方法相比,更适合工业化生产。
[0116]
下面结合具体实施,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
[0117]
实施例1
[0118][0119]
在反应瓶中加入二氯甲烷(100ml),将式ⅰ化合物(10.0g,0.062mol)加入反应瓶中,搅拌溶解,加入无水三氯化铝(12.4g,0.093mol),连接尾气管,做好尾气吸收,搅拌均匀,滴加吡啶(5.9g,0.0744mol),升温至40℃反应2h,点板确认反应进度,反应完毕,降温至20℃,冰浴降温至0℃,滴加纯化水(100ml),控温15-20℃之间,搅拌20min,静置分层,取有机相,无水硫酸钠干燥,旋蒸除去溶剂,得到类白色固体(10.8g),收率:86.2%(相对于式ⅰ)。ms(esi):[m 1]
=205.27。
[0120]
实施例2
[0121][0122]
将式ⅲ化合物(15.0g,0.073mol)加入硫酸(80ml)中,搅拌均匀,冰盐降温至0℃,保持搅拌,配制硝酸(9.4ml,0.146mol)/硫酸(9.4ml)1:1溶液(浓硝酸(68%),浓硫酸(98%)),保温0℃滴加进入反应液中,保温0℃反应1h,将反应液倒入冰块中淬灭,加入二氯甲烷萃取,收集有机相,纯化水和饱和食盐水各洗涤一遍,无水硫酸钠干燥,过滤,蒸馏除去溶剂,得到式ⅳ化合物(17.1g),收率:93.7%(相对于式ⅲ)。ms(esi):[m 1]
=250.27。
[0123]
实施例3
[0124][0125]
将式ⅳ化合物(50.0g,0.20mol)加入甲醇(450ml)水(50ml)中,氮气置换3次,氮气保护下加入10%钯炭(4g),搅拌均匀,通入氢气,气压0.5mpa,保温25℃反应4h,过滤除去钯炭,蒸馏得到化合物
ⅴ
(39.3g),收率:89.3%(相对于式ⅳ)。ms(esi):[m 1]
=220.28。
[0126]
实施例4
[0127][0128]
将式
ⅴ
化合物(50.0g,0.23mol)加入无水四氢呋喃(500ml)中,搅拌控温0-5℃,加入式ⅵ化合物(49.2g,0.25mol),加入三乙胺(27.9g,0.28mol)保温0-5℃反应4.5h,反应完毕,滴加1.0mhcl调节ph至酸性,有大量固体析出,过滤得到粗品式ⅶ化合物(85.6g),加入乙腈(500ml),重结晶,降温至0℃搅拌析晶1h,过滤得到纯化式ⅶ产物(77.5g)收率:90.1%(相对于式
ⅴ
)。ms(esi):[m 1]
=378.36。
[0129]
实施例5
[0130][0131]
在反应瓶中加入二氯甲烷(100ml),将式ⅰ化合物(10.0g,0.062mol)加入反应瓶中,搅拌溶解,加入无水三氯化铝(12.4g,0.093mol),连接尾气管,做好尾气吸收,搅拌均匀,滴加吡啶(6.9g,0.0868mol),升温至40℃反应2h,点板确认反应进度,反应完毕,降温至20℃,冰浴降温至0℃,滴加纯化水(100ml),控温15-20℃之间,搅拌20min,静置分层,取有机相,无水硫酸钠干燥,旋蒸除去溶剂,得到类白色固体(10.0g),收率:84.9%(相对于式ⅰ)。ms(esi):[m 1]
=205.27。
[0132]
实施例6
[0133][0134]
在反应瓶中加入二氯甲烷(100ml),将式ⅰ化合物(10.0g,0.062mol)加入反应瓶中,搅拌溶解,加入无水三氯化铝(12.4g,0.093mol),连接尾气管,做好尾气吸收,搅拌均匀,滴加吡啶(5.9g,0.0744mol),升温至40℃反应2h,点板确认反应进度,反应完毕,降温至20℃,冰浴降温至0℃,滴加纯化水(100ml),控温30℃左右,搅拌20min,静置分层,取有机相,无水硫酸钠干燥,旋蒸除去溶剂,得到类白色固体(8.8g),收率:69.5%(相对于式ⅰ)。ms(esi):[m 1]
=205.27。
[0135]
实施例7
[0136][0137]
将式ⅳ化合物(50.0g,0.20mol)加入甲醇(450ml)水(50ml)中,氮气置换3次,氮气保护下加入8%钯炭(5g),搅拌均匀,通入氢气,气压0.5mpa,保温25℃反应4h,过滤除去钯炭,蒸馏得到化合物
ⅴ
(32.6g),收率:74.1%(相对于式ⅳ)。ms(esi):[m 1]
=220.28。
[0138]
实施例8
[0139][0140]
将式ⅳ化合物(50.0g,0.20mol)加入甲醇(450ml)水(50ml)中,氮气置换3次,氮气保护下加入5%钯炭(10g),搅拌均匀,通入氢气,气压0.5mpa,保温25℃反应4h,过滤除去钯炭,蒸馏得到化合物
ⅴ
(28.1g),收率:63.9%(相对于式ⅳ)。ms(esi):[m 1]
=220.28。
[0141]
实施例9
[0142][0143]
将式ⅳ化合物(50.0g,0.20mol)加入甲醇(450ml)水(50ml)中,氮气置换3次,氮气保护下加入10%钯炭(4g),搅拌均匀,通入氢气,气压1mpa,保温25℃反应4h,过滤除去钯炭,蒸馏得到化合物
ⅴ
(30.1g),收率:68.4%(相对于式ⅳ)。ms(esi):[m 1]
=220.28。
[0144]
实施例10
[0145][0146]
将式ⅳ化合物(50.0g,0.20mol)加入甲醇(450ml)水(50ml)中,氮气置换3次,氮气保护下加入10%钯炭(4g),搅拌均匀,通入氢气,气压0.2mpa,保温25℃反应4h,过滤除去钯炭,蒸馏得到化合物
ⅴ
(37.6g),收率:85.5%(相对于式ⅳ)。ms(esi):[m 1]
=220.28。
[0147]
实施例11
[0148][0149]
将式ⅳ化合物(50.0g,0.20mol)加入甲醇(500ml),氮气置换3次,氮气保护下加入10%钯炭(4g),搅拌均匀,通入氢气,气压0.2mpa,保温25℃反应4h,过滤除去钯碳,蒸馏得到化合物
ⅴ
(22.1g),收率:50.3%(相对于式ⅳ)。ms(esi):[m 1]
=220.28。
[0150]
实施例12
[0151][0152]
将式ⅳ化合物(50.0g,0.20mol)加入甲醇(350ml)水(150ml)中,氮气置换3次,氮气保护下加入10%钯碳(4g),搅拌均匀,通入氢气,气压0.2mpa,保温25℃反应4h,过滤除去
钯碳,蒸馏得到化合物
ⅴ
(8.8g),收率:20.1%(相对于式ⅳ)。ms(esi):[m 1]
=220.28。
[0153]
综上,本发明的方法可以在低温下实现式v化合物的简单高效合成,且产率高。从而可以低成本的合成拉司米地坦。
[0154]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。