三(4-乙炔苯基)胺类共轭微孔聚合物、制备方法及应用
技术领域
1.本发明属于聚合物制备技术领域,具体涉及一种三(4-乙炔苯基)胺类共轭微孔聚合物、制备方法及应用。
背景技术:
2.运用太阳能将水转换为清洁燃料氢气,是解决当前能源危机与环境污染的理想途径之一。这是由于太阳能具有来源丰富、清洁环保、取之不尽用之不竭的特点,同时,氢气热值高,燃烧后生成无环境污染的水又可以循环作为制备氢气的原料,但该过程的实用化有赖于高性能催化剂的开发和利用。作为催化剂的一种,共轭微孔聚合物(cmps)由于光吸收率高、稳定性好、来源广泛、价格低廉、合成方法多样、结构和能级水平易于调控等优势引起了人们广泛的研究兴趣。
3.自从2015年cooper等首次报道了cmps的光催化水分解性能以来,共轭微孔聚合物的光催化性能研究取得了巨大的进展。研究已经发现聚合物的构筑单元、构筑单元的相对浓度和连接位置是影响聚合物光催化性能的重要因素。具有供体-给体(d-a)结构的cmps尤其是含有n-和s-的受体单元与大共轭结构的供体单元构筑的cmps通常具有较高的催化活性,这可能是由于该结构的聚合物具有较宽的吸收光谱,因而具有较高的太阳光吸收效率,能够增强分子内偶极极化,加速电荷转移,改善截面湿润性。另外,由于三键和双键的引入可以进一步扩展聚合物的共轭结构,增强载流子的迁移率,因而可以进一步改善材料的光催化性能。然而,目前已报道的三官能团及以上cmps类光催化剂构筑单元本就有限,具有三键的多官能团的单体更是鲜有报道,极大的限制了cmps类光催化剂的研究进展。
4.三(4-乙炔苯基)胺是一种常见的具有三键的三官能团供体单元,含有该单元的共轭聚合物材料已经在光电领域、吸附领域等显示出优异的潜在价值,对其在光催化领域的应用进行探索,对开发新型的多官能团单体有利。苯并噻二唑单元由于具有优异的受电子性能和电化学稳定性与可逆性,同时含有n-和s-原子,已被广泛应用于光催化水分解cmps的构筑中,并显示出良好的光催化性能,进一步探索苯并噻二唑单元连接位置对材料光催化性能的影响,将对新型光催化剂的设计合成提供理论依据。
技术实现要素:
5.针对现有技术中存在的问题,本发明提供了一种三(4-乙炔苯基)胺类共轭微孔聚合物,以三(4-乙炔苯)胺(tea)为三官能团单体,以不同连接位置的苯并噻二唑(bt)单元为双官能团单体,通过钯催化的sonogashira-hagihara交叉偶联反应,合成四种基于三(4-乙炔苯基)胺单元和不同连接位置的苯并噻二唑单元的d-a结构的cmps。
6.本发明的另一目的是提供了三(4-乙炔苯基)胺类共轭微孔聚合物在可见光下光催化水分解中的应用,研究发现tea是一种潜在性能优异的多官能团构筑单体,bt单元的连接位置可以有效调控聚合物的光催化析氢性能。
7.为解决上述技术问题,本发明采用以下技术方案:
8.本发明三(4-乙炔苯基)胺类共轭微孔聚合物的制备方法,通过三(4-乙炔苯基)胺m0和具有不同连接位置的苯并噻二唑单体m1、m2、m3和m4聚合制备,具体包括以下步骤:氩气状态下,往史莱克反应瓶中加入三(4-乙炔苯基)胺m0、苯并噻二唑单体、四(三苯基膦)钯、碘化亚铜、无水dmf和无水三乙胺,密封反应,反应结束后自然冷却至室温,过滤,所得固体洗涤、真空干燥得三(4-乙炔苯基)胺类共轭微孔聚合物;合成路线如下所示:
[0009][0010]
进一步,所述三(4-乙炔苯基)胺m0与苯并噻二唑单体的摩尔比为1:1。
[0011]
进一步,所述m0、四(三苯基膦)钯和碘化亚铜的摩尔比为1:0.06:0.2。
[0012]
进一步,无水dmf和无水三乙胺的体积比为1:1,以0.5mol三(4-乙炔苯基)胺为基准,需要无水dmf5 ml。
[0013]
进一步,密封反应是在100℃条件下搅拌反应72h。
[0014]
进一步,所述真空干燥温度为60℃,干燥时间为24h。
[0015]
利用本发明所述的制备方法制得的三(4-乙炔苯基)胺类共轭微孔聚合物。
[0016]
本发明所述的三(4-乙炔苯基)胺类共轭微孔聚合物在光催化水分解中的应用,所述三(4-乙炔苯基)胺类共轭微孔聚合物的结构式如下所示:
[0017][0018]
进一步,改变苯并噻二唑单元的连接位置,可以有效的调控材料的析氢性能,随着聚合物中苯并噻二唑单元的连接位置由5,6-位
→
4,7-位的变化,聚合物的析氢效率逐渐增大,fs4具有最高的析氢效率,高达115.74μmol g-1
h-1
,为fs1的三倍。
[0019]
本发明的有益效果:本发明通过sonogashira-hagihara交叉偶联反应合成了四种基于三(4-乙炔苯基)胺单元和苯并噻二唑单元的共轭微孔聚合物fs1、fs2、fs3和fs4,聚合物中苯并噻二唑单元的连接位置分别为:5,6-位、4,5-位、4,6-位和4,7-位。可见光驱动下,四种聚合物均具有稳定的光催化析氢性能,三(4-乙炔苯基)胺单元是一种潜在的用于光催化水分解领域的共轭微孔聚合物材料多官能团构筑单体。4,7-位连接的聚合物fs4性能最好,其析氢效率高达115.74μmol g-1
h-1
,为fs1的近三倍。苯并噻二唑单元连接位置可以有效调控聚合物的性能。
附图说明
[0020]
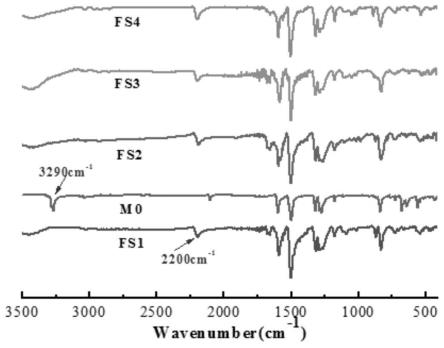
图1为m0及聚合物的红外光谱图;
[0021]
图2为聚合物的热失重曲线;
[0022]
图3为聚合物的粉末x衍射图;
[0023]
图4为聚合物的扫描电镜照片;
[0024]
图5为吸附-脱附性能和孔径分布图,其中(a)聚合物的氮气吸附-脱附等温曲线;(b)孔径分布图;
[0025]
图6为聚合物的紫外-可见吸收光谱和光致发光光谱,其中(a)聚合物的紫外-可见吸收光谱;(b)聚合物的光致发光光谱;
[0026]
图7为聚合物的循环伏安曲线;
[0027]
图8为聚合物的homo和lumo的能带位置;
[0028]
图9聚合物的光催化水分解产氢量与时间的关系图和fs4的循环稳定性,其中(a)聚合物的光催化水分解产氢量与时间的关系图(λ》420nm);(b)fs4的循环稳定性;
[0029]
图10为聚合物的光电流随时间变化曲线图和电化学交流阻抗图(eis),其中(a)聚合物的光电流随时间变化曲线图;(b)电化学交流阻抗图(eis)。
具体实施方式
[0030]
下面结合具体实施例,对本发明做进一步说明。应理解,以下实施例仅用于说明本发明而非用于限制本发明的范围,该领域的技术熟练人员可以根据上述发明的内容作出一些非本质的改进和调整。
[0031]
三(4-乙炔苯基)胺(m0,w=97%):分析纯,郑州阿尔法化工有限公司;4,7-二溴-2,1,3-苯并噻二唑(m4,w=99%):分析纯,深圳睿迅光电材料科技有限公司;四(三苯基膦)钯(w=99%):分析纯,stream公司;碘化亚铜(w=99%):分析纯,北京伊诺凯科技有限公司;n,n-二甲基甲酰胺(w=99.5%):分析纯,天津市富裕精细化工有限公司;三乙胺(w=99%):分析纯,天津市科密欧化学试剂有限公司。
[0032]
聚合物由三(4-乙炔苯基)胺m0和具有不同连接位置的苯并噻二唑单体m1、m2、m3和m4聚合制备。具体的合成路线如下所示。
[0033][0034]
实施例1
[0035]
本实施例聚合物fs1的合成方法如下:
[0036]
氩气状态下,往带有搅拌子的120ml干燥的史莱克反应瓶中加入m0(158.9mg,0.5mmol)、m1(147.3mg,0.5mmol)、四(三苯基膦)钯(40.8mg,0.03mmol)、碘化亚铜(20.5mg,0.10mmol)、无水dmf(5ml)和无水三乙胺(5ml),密封,100℃条件下搅拌反应72h。自然冷却至室温,得含有浅红黑色固体的混合物。过滤,所得固体依次用氯仿、水、甲醇、丙酮洗涤,再依次以氯仿、甲醇为溶剂,索氏提取器提取,60℃真空干燥24h,得220.0mg浅黑红色固体粉末fs1,产率:98%.ft-ir(kbr,cm-1
):2194,1588,1496,1310,1116,828.anal.calcd for c
30h14
n3s:c,80.34;h,3.15;9.37.found:c,75.32;h,3.77;n,7.91%.元素分析理论值和测定值的偏离可能是由于聚合物端基残留引起的,该现象在聚合物元素分析测试中常见。
[0037]
5,6-二溴-2,1,3-苯并噻二唑(m1)的合成方法如下:
[0038]
无水无氧条件下,往含有邻苯二胺(21.6g,0.2mol),吡啶(200ml)的澄清溶液中于0℃下分批加入对甲苯磺酰氯(120g,0.63mol),加完后室温反应2day,将反应液倒入冰水中搅拌,过滤,水洗,盐酸(800ml,1m)洗,水洗,真空干燥,紫外烘干,共得到产物n,n
’‑
二(4-甲基苯磺酰胺)邻苯二胺76.062g,产率:91%。
[0039]1h nmr(300mhz,cdcl3):δ7.58(d,j=8.7hz,4h),7.21(d,j=8.7hz,4h),7.10(s,broad peak,2h),7.00(m,4h),2.39(s,6h)。
[0040]
往含有(n,n
’‑
二(4-甲基苯磺酰胺)邻苯二胺(76.26g,0.183mol),醋酸钠(30.50g,0.372mol),乙酸(305ml)的反应混合物中逐滴加入溴(18.9ml,0.367mol),1.5h加完,室温反应1h,然后加热至100℃反应1h,停止反应,将反应液倒入900ml水中,过滤水洗,醋酸重结晶三次,得n,n
’‑
二(4-甲基苯磺酰胺)-5,6-二溴-邻苯二胺的粗产物80.56g,产率:77%。
[0041]1h nmr(300mhz,cdcl3):δ7.59(d,j=8.7,4h),7.27(d,j=8.7,4h),7.20(s,2h),6.86(s,1h),2.43(s,6h).
[0042]
n,n
’‑
二(4-甲基苯磺酰胺)-5,6-二溴-邻苯二胺(80.56g,0.140mol),硫酸(98%,161ml)和水(8.0ml)于40℃下搅拌反应约12h,将反应液倒入冰水中,用50%氢氧化钠溶液中和至ph值为14,过滤,滤饼用乙醇/水重结晶,得红色固体产物4,5-二溴-邻苯二胺33.10g,产率89%。
[0043]1h nmr(300mhz,cdcl3):δ6.93(s,2h),3.42(s,4h)
[0044]
无水无氧条件下,在含有5,6-二溴邻苯二胺(33.10g,0.124mol),吡啶(62ml)的溶液中,于0℃下逐滴加入二氯亚砜(62ml),室温反应4day,将反应液逐滴加入到冰水中,然后加硅藻土过滤,滤饼充分用二氯甲烷洗涤,滤液用二氯甲烷萃取,有机相合并,饱和食盐水洗,无水硫酸钠干燥,柱层析分离,淋洗剂:石油醚:二氯甲烷=6:1,所得产物用丙酮重结晶,得到白色固体产物5,6-二溴-2,1,3-苯并噻二唑24.783g,产率68%。ms-ei:294(m
).1h nmr(300mhz,cdcl3):δ8.3(s,2h).
13
c nmr(300mhz,cdcl3):δ153.78,127.21,124.92.anal.calcd for c6h2br2n2s:c,24.51%;h,0.69%;n,9.53%;s,10.91%;br,54.36%.found:c,24.64%;h,0.67%;n,9.55%;s,10.81%;br,54.22%.m.p.135-136℃.
[0045]
实施例2
[0046]
本实施例聚合物fs2的合成方法如下:
[0047]
fs2采用与fs1相同的合成方法。
[0048]
m0(158.6mg,0.5mmol)、m2(145.3mg,0.5mmol)、四(三苯基膦)钯(40.3mg,0.03mmol)、碘化亚铜(21.0mg,0.1mmol),得210.0mg黑黄色固体粉末fs2,产率:94%.ft-ir(kbr,cm-1)
:2189,1595,1496,1314,1182,833.anal.calcd for c
30h14
n3s:c,80.34;h,3.15;9.37.found:c,72.96;h,3.96;n,8.55%.
[0049]
4,5-二溴-2,1,3-苯并噻二唑(m2)的合成方法如下:
[0050]
在含有二水合氯化亚锡(120g,0.53mol),盐酸(220ml)的溶液中,分批加入4-溴-6-硝基苯胺,约2h加完,加完后反应液于55℃下反应10h,将反应液倒入冰水中,氢氧化钠中和至ph值为14,真空干燥过夜,直接进行下一步反应。
[0051]
将上一步所得到的产物溶于吡啶(50ml)中,于0℃下逐滴加入二氯亚砜(100ml),
加完后室温反应3天,将反应液逐滴加入到冰水中,加硅藻土过滤,滤饼充分用二氯甲烷洗涤,滤液二氯甲烷洗,有机相合并用饱和食盐水洗,无水硫酸钠干燥,柱层析分离,淋洗剂:石油醚:二氯甲烷=6:1,得到白色固体产物5-溴-2,1,3-苯并噻二唑15.168g,产率:59%。1h nmr(300mhz,cdcl3):δ8.23(d,j1=1.2hz,1h),7.88(d,j2=9.3hz,1h),7.67(dd,j1=1.2hz,j2=9.3hz,1h)。
[0052]
在5-溴-2,1,3-苯并噻二唑(5.3g,20mmol),氢溴酸(40%,28ml)的澄清溶液中,于100℃下逐滴加入溴(1.2ml),加完后在此温度下反应2h,然后再滴加溴(1.2ml),加完后继续反应4h,停止反应,将反应液倒入饱和亚硫酸氢钠的水溶液中,氯仿萃取,有机相合并,饱和食盐水洗,无水硫酸钠干燥,柱层析分离,淋洗剂:石油醚:二氯甲烷=6:1,得到白色固体:5.338g,产率:91%。
[0053]
ms-ei:294(m
).1h nmr(300mhz,cdcl3):δ7.84(d,j=9.6hz,1h),7.79(d,j=9.6hz,1h);
13
c nmr(300mhz,cdcl3):δ154.34,152.97,134.20,127.44,120.73,117.09.anal.calcd for c6h2br2n2s:c,24.51%;h,0.69%;n,9.53%;s,10.91%br,%54.36.found:c,24.94%;h,0.63%;n,9.79%;br,54.65%;s,10.85%.m.p.138-139℃.
[0054]
实施例3
[0055]
本实施例聚合物fs3的合成方法如下:
[0056]
fs3采用与fs1相同的合成方法。
[0057]
m0(157.8mg,0.5mmol)、m3(145.8mg,0.5mmol)、四(三苯基膦)钯(40.5mg,0.03mmol)、碘化亚铜(21.5mg,0.1mmol)得210.0mg浅棕色固体粉末fs3,产率:94%.ft-ir(kbr,cm-1
):3026,2199,1582,1500,1315,1093,825.anal.calcd for c
30h14
n3s:c,80.34;h,3.15;9.37.found:c,71.48;h,3.92;n,7.80%.
[0058]
4,6-二溴-2,1,3-苯并噻二唑(m3)的合成方法如下:
[0059]
在250ml反应瓶中,加入邻硝基苯胺(20g,0.145mol),乙酸(100ml),将此反应混合物加热到55℃使邻硝基苯胺完全溶解。在40min内分批往反应混合物中加入nbs(77.391g,0.435mol),将反应液温度降到40-50℃并在此温度范围内反应3h,停止反应。将反应液倒入1.7l冰水中,过滤,水洗,乙醇重结晶,抽干,共得到红色针状固体产物4,6-二溴-2-硝基苯胺37.459g,产率87%。1h nmr(300mhz,cdcl3):δ8.29(d,j=2.4hz,1h),7.81(d,j=2.4hz,1h),6.64(s,2h).
[0060]
往含有二水合氯化亚锡(64g,0.281mol),盐酸(115ml)的溶液中分批加入2,4-二溴-6-硝基苯胺(18.5g,0.063mol),加完后室温反应10min,然后加热至70℃反应1h,待红色针状固体全部消失,在室温下再反应1h,将反应液倒入冰水中,氢氧化钠调节ph值为14,过滤,真空干燥,得产物4,6-二溴邻苯二胺16.051g。产率97%。
[0061]1h nmr(300mhz,cdcl3):δ7.10(d,j=2.1hz,1h),6.78(d,j=2.1hz,1h),3.63(s,broad peak,4h).
[0062]
无水无氧条件下,往4,6-二溴邻苯二胺(16.051g,0.060mol)的吡啶(49ml)溶液中,于0℃下逐滴加入二氯亚砜(49ml),加完后室温反应3.5day。将反应液倒入冰水中,用硅藻土过滤,滤饼充分用二氯甲烷洗,分离有机相,有机相用饱和食盐水洗,无水硫酸钠干燥,柱层析分离,淋洗剂:石油醚:二氯甲烷=6:1,得产物14.174g,产率:80%。取一部分用乙酸乙酯重结晶,重结晶后得到白色针状晶体4,6-二溴-2,1,3-苯并噻二唑3.8g。
[0063]
ms-ei:294(m
).1h nmr(300mhz,cdcl3):δ8.18(d,j=2.1,1h),7.95(d,j=2.1,1h).
13
c nmr(300mhz,cdcl3):δ154.68,152.31,135.22,124.20,123.17,114.99.anal.calcd for c6h2br2n2s:c,24.51%;h,0.69%;n,9.53%;s,10.91%;br,54.36%.found:c,24.52%;h,0.71%;n,9.52%;s,10.97%;br,54.30%.m.p.127-128℃.
[0064]
实施例4
[0065]
本实施例聚合物fs4的合成方法如下:
[0066]
fs4采用与fs1相同的合成方法。
[0067]
m0(158.6mg,0.50mmol)、m4(146.8mg,0.5mmol)、四(三苯基膦)钯(40.3mg,0.03mmol)、碘化亚铜(20.4mg,0.1mmol)得212.3mg砖红色固体粉末fs4,产率:,95%.ft-ir(kbr,cm-1
):3034,2202,1598,1499,1315,1169,830.anal.calcd for c
30h14
n3s:c,80.34;h,3.15;9.37.found:c,66.70;h,3.80;n,7.76%.
[0068]
一、测试与表征
[0069]
核磁共振波谱(1h nmr和
13
c nmr)分析仪(瑞士bruker公司am-300):采用cdcl3为溶剂,tms为内标测定;傅里叶变换红外光谱仪(美国thermo scientific公司nicolet is50):采用kbr压片法,kbr:样品=150:1(weight:weight)放入玛瑙研钵中研磨均匀,置于压模机中,压成透明的薄片样品,扫描范围4000-500cm-1
;全自动元素分析仪(德国elementar公司elemantar varioel elⅲ);x射线光电子能谱仪(美国thermo fisher scientific公司escalab250xi);热失重分析仪(德国netzsch公司netzsch sta449 f3):氮气氛围下测定,升温速率10℃/min,升温范围为25℃-750℃;场发射扫描电子显微镜(日本jeol公司jsm-7001f);x射线衍射仪(德国bruker公司d8x);紫外可见光谱仪(日本hitachi公司u-3900):以硫酸钡固体圆片做背景,测试时样品铺满样品槽表面,扫描速率:120nm/min,扫描范围:800nm-200nm;荧光光谱仪(日本hitachi公司f7000):激发波长360nm;气体吸附仪(美国micromeritics公司asap 2460):77k下氮气吸附-脱附测试,样品称重范围:80mg-120mg,测试前在120℃下真空脱气12h;聚合物的孔性能分析采用brunauer-emmet-teller(bet)算法;聚合物的孔尺寸及其分布曲线采用非定域密度泛函理论(nldft)模型分析;电化学工作站(上海辰华公司chi660d):采用三电极体系,样品浓度1mg/ml,用乙醇稀释超声分散,移液枪吸取12μl的样品滴加到工作电极上,晾干测定,ag/agcl电极为参比电极,pt片为对电极;电化学性能测定时以玻碳电极为工作电极,电解液为0.1mol/l[bu4n]clo4的乙腈溶液,扫描范围-1v-1v,扫描速度100mv/s。聚合物的最高占有轨道(homo)通过公式计算,最低未占有轨道(lumo)按公式计算,其中为氧化起始电位,为还原起始电位;光电流测定时将0.1mol的na2so4配制成250ml的水溶液作为电解液,工作电极采用ito导电玻璃,40s的光照周期;电化学阻抗谱(eis)测定时:玻碳电极为工作电极,将1.8638g mol kcl(0.1mmol),0.5280g k4fe(cn)6·
3h2o(5mmol)和0.4115g k3fe(cn)6(5mmol)配制成250ml的水溶液作为电解液。
[0070]
1、聚合物结构分析
[0071]
聚合物fs1、fs2、fs3和fs4均为不溶不熔的粉末,溶剂测试均不溶于一般溶剂如水、氯仿、甲醇、丙酮等,在一般的酸或碱的水溶液中也不发生变化,说明聚合物具有良好的
溶剂稳定性和化学稳定性,这可能是由于聚合物的刚性共轭结构和高度交联的网络结构引起的。
[0072]
单体m0的红外光谱图(图1)中可以清晰的看到3290cm-1
处端基三键上c(sp)-h键的伸缩振动峰,2200cm-1
附近c(sp)-c(sp2)-的弱信号峰,但在聚合物的红外光谱(图1)中,3290cm-1
处的特征峰消失,2200cm-1
附近峰的信号明显增强,说明端基三键上c-h键参与反应,形成了c(sp)-c(sp2)-,聚合反应成功。聚合物的x射线光电子能谱(xps)测试结果中溴元素的信号几乎为零,进一步说明了聚合反应的发生。
[0073]
聚合物的热失重分析结果如图2。氮气环境下,fs1、fs2、fs3和fs4的10%热失重分解温度分别为450℃、470℃、517℃、517℃,温度逐渐升高,说明随着聚合物中苯并噻二唑单元连接位置由5,6-位
→
4,7-位的变化,聚合物的热稳定性能逐渐提高,同时所有的聚合物在600℃时仍然保持50%的残余量,表明四种聚合物均具有良好的热稳定性。
[0074]
聚合物fs1、fs2、fs3和fs4的粉末x射线衍射谱图(图3)相似,2theta值在15-20
°
范围内出现一个大包峰,无明显的尖峰存在,说明聚合物均为无定型结构。聚合物的扫描电镜(图4)结果显示四种共轭微孔聚合物均呈现大小均一的纳米球状颗粒聚集形态,形貌相近,说明聚合物中苯并噻二唑单元的连接位置对聚合物的形貌影响较小。
[0075]
2、聚合物孔性能分析
[0076]
聚合物孔性能通过氮气吸附-脱附实验获得。先将聚合物于120℃下真空干燥12h,后于77k下测定聚合物的氮气吸附-脱附曲线(图5(a))。四种聚合物均表现为ⅰ型吸附曲线特征,吸附量fs4》fs3》fs2》fs1,并且差异明显;在相对低压区(p/p0《0.01),四种聚合物对氮气的吸附量均有陡升趋势,但明显程度fs4》fs3》fs2》fs1,说明聚合物均具有微孔,但微孔体积fs4》fs3》fs2》fs1;在相对高压区(p/p0》0.2),四种聚合物对氮气的吸附量均上升缓慢,表明聚合物中均存在介孔和大孔。根据brunauer-emmet-teller(bet)算法计算出聚合物的比表面积和多孔性质如表1。聚合物fs1、fs2、fs3和fs4的比表面积分别为5.2m2·
g-1
、11.86m2·
g-1
、89.95m2·
g-1
和367.57m2·
g-1
,说明随着苯并噻二唑单元连接位置由5,6-位
→
4,7-位的变化,聚合物的比表面积逐渐增大,大的比表面积通常意味着良好的光催化水分解性能。聚合物的孔尺寸以及其分布曲线采用非定域密度泛函理论(nldft)模型分析获得(图5(b))。四种聚合物主要以微孔存在,介孔和大孔较少,fs4的孔径分布相对来说较宽,微孔分布较多,同时也存在一定量的介孔和大孔结构。四种聚合物均由相同的结构单元构成,因而聚合物孔性能的变化只能是由聚合物中苯并噻二唑单元连接位置的变化引起的,这一结果说明,聚合单元的连接位置也是影响聚合物孔性能的重要因素,调控聚合单元在聚合物中的连接位置,可以有效调控聚合物的孔性能。
[0077]
表1聚合物的比表面积和多孔性质
[0078][0079]
3.紫外-可见吸收光谱和荧光光谱分析
[0080]
聚合物的紫外-可见吸收光谱和荧光光谱如图6。四种聚合物的吸收光谱都拓展到近700nm,几乎涵盖整个可见光区域,表明这些聚合物能够充分吸收太阳光,聚合物的这一性能对提高太阳光的利用效率有利。但聚合物的吸收边不同,fs1、fs2、fs3和fs4的吸收边分别为531nm、536nm、513nm、558nm,fs4的吸收边明显红移,这主要是由于聚合物fs4中苯并噻二唑单元的4,7-位连接方式提高了聚合物骨架的共平面性,增大了聚合物的共轭程度;聚合物fs4吸光能力最强,暗示fs4可能具有最高的光催化活性。根据吸收边计算出fs1、fs2、fs3和fs4的光学带宽分别为1.83ev、1.79ev、2.06ev、1.74ev,均具有较窄的带隙。随着聚合物中苯并噻二唑单元连接位置由5,6-位
→
4,7-位的变化,聚合物的发光强度逐渐减弱,在相同的测试条件下,fs4的发光强度最低,这可能是聚合物的共轭度增加对荧光的猝灭作用引起的,说明其可能具有最佳的光生电子-空穴分离能力,较多的载流子参与催化剂表面的氧化还原反应,有利于提高析氢性能。
[0081]
4.电化学性能分析
[0082]
聚合物的循环伏安曲线(图7)在负区均呈现苯并噻二唑单元的明显的可逆的还原和去还原峰,正区可以看到属于三苯胺的氧化峰,但去氧化峰不明显。由循环伏安曲线,可以计算出四种聚合物的homo和lumo值,进一步计算出电化学带宽,详细数据如表2。fs1、fs2、fs3和fs4的lumo能级分别为-3.78ev、-3.81ev、-3.76ev和-3.83ev,均高于h2o/h2的还原电势(-4.5ev)(图8),表明聚合物均能提供足够的热力学驱动力分解水制氢。
[0083]
表2聚合物的光物理和光催化性能
[0084][0085]a聚合物的电化学宽带
[0086]b聚合物的光学带宽
[0087]
二、可见光驱动光催化水分解制氢
[0088]
往带有搅拌子的80ml特制的石英测试管中,加入20ml水与三乙醇胺(v:v=4:1)的混合溶剂,将20mg的光催化剂分散到混合溶剂中,三乙醇胺作为牺牲剂。密封塞密封,往测试管中通入高纯氩气鼓泡0.5h以排出体系内的空气,超声0.5h将催化剂均匀的分散于混合溶剂内。将分散后的测试管放到密闭反应器中,300w氙灯作为光源,光照下搅拌,进行光催化水分解反应,通入循环冷却水保持反应温度为25℃,每小时用注射器抽取0.4ml的气体通过装有tcd的安捷伦7890a气相色谱仪在线监测析氢速率。析氢速率(单位:μmolg-1
h-1
)采用下式计算。
[0089][0090]
式中:峰面积:gc检出峰的峰面积
[0091]
9.39494
×
10-11
:换算因子
[0092]
68
×
106:反应容器内空气体积
[0093]
0.4:进样体积,单位:ml
[0094]
0.02:样品质量,单位:g
[0095]
以三乙醇胺作为牺牲剂时可见光驱动下聚合物的光催化水分解析氢量与时间的关系如图9。聚合物均表现出稳定的析氢效果。相同条件下聚合物fs1、fs2、fs3和fs4的析氢效率分别为34.93μmol g-1
h-1
、45.58μmol g-1
h-1
、90.43μmol g-1
h-1
和115.74μmol g-1
h-1
,说明三(4-乙炔苯基)胺类共轭微孔聚合物具有良好的光催化水分解性能,三(4-乙炔苯基)胺可能是一种性能优异的共轭微孔聚合物类光催化剂的多官能团构筑单元;同时,随着聚合物中苯并噻二唑单元的连接位置由5,6-位
→
4,7-位的变化,聚合物的析氢效率逐渐增大,fs4具有最高的析氢效率,高达115.74μmol g-1
h-1
,为fs1的三倍,说明改变苯并噻二唑单元的连接位置,可以有效的调控材料的析氢性能。
[0096]
进一步测试了性能最佳的聚合物fs4的结构稳定性和循环稳定性,光催化前后fs4的红外光谱几乎没有发生改变,说明fs4具有良好的结构稳定性。通过测定每小时析氢量,研究了fs4的析氢循环稳定性(图9)。连续18h光照下,循环三次,聚合物的析氢效率没有明显的变化,表明fs4具有优异的循环稳定性。
[0097]
为了评价聚合物的电荷分离能力,进行了8个周期的瞬态光电流响应测试(图10)。fs4的光电流最高,电流密度最大,即具有良好的光致电子-空穴对分离能力。聚合物的界面电子转移行为通过电化学交流阻抗(eis)测试,结果显示聚合物fs4电弧半径最小,说明fs4的电化学电阻最小,即fs4的电子导电性和电荷转移相对较强,进一步验证了fs4具有较强的电子-空穴分离能力,这与光电流响应结果相一致,也与四种聚合物的析氢速率一致。
[0098]
以上显示和描述了本发明的基本原理和主要特征以及本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。