喹啉化合物的晶型
ɑ
及其制备方法和应用
技术领域
1.本发明涉及晶型制备技术领域,具体涉及喹啉化合物的晶型
ɑ
及其制备方法和应用。
背景技术:
2.喹啉类化合物常被用于治疗恶性肿瘤,例如中国专利文献cn102977014a在说明书中记载了具有式i的化合物及其盐,说明书中记载了其可用于抑制肺癌细胞、结肠癌细胞、胃癌细胞、肝癌细胞、乳腺癌细胞、恶性胶质母细胞瘤细胞等多种肿瘤细胞。
[0003][0004]
本发明人根据中国授权专利文献cn102977014a所公开的方法制备式(i)所示的化合物并进行分析的结果,发现所获得的生成物具有较差的稳定性,而且生物利用度低下,体内吸收效果较差,不适合用作医药原料。
技术实现要素:
[0005]
因此,本发明的目的在于提供上述式(i)所示的喹啉化合物的晶型
ɑ
及其制备方法和用途,该晶型
ɑ
具有良好的稳定性,且在药代动力学实验中具有明显提高的生物利用度。
[0006]
本发明提供了下述式(i)所示的喹啉化合物的晶型
ɑ
,以2θ角度表示的x-射线粉末衍射,在3.8
±
0.2
°
、7.6
±
0.2
°
处有特征峰。
[0007][0008]
进一步地,以2θ角度表示的x-射线粉末衍射,还在18.0
±
0.2
°
和/或26.4
±
0.2
°
处有特征峰。
[0009]
进一步地,以2θ角度表示的x-射线粉末衍射,还在14.0
±
0.2
°
、17.3
±
0.2
°
、22.8
±
0.2
°
、23.8
±
0.2
°
处有特征峰。
[0010]
进一步地,所述晶型
ɑ
的xrpd图谱数据为:
[0011]
峰编号2θ(
°
)相对强度(%)峰编号2θ(
°
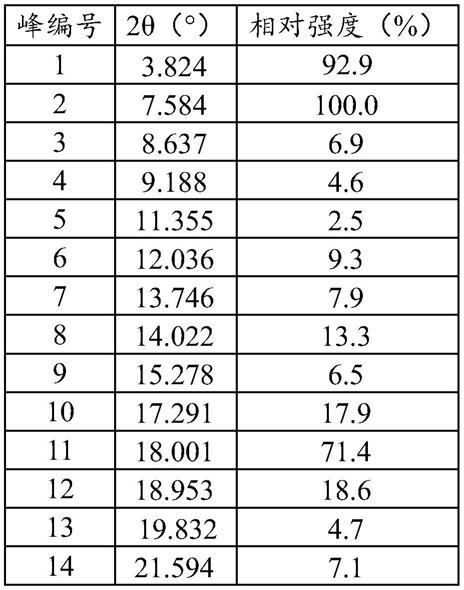
)相对强度(%)13.82492.91218.95318.627.584100.01319.8324.738.6376.91421.5947.149.1884.61522.78316.9511.3552.51623.2939.2612.0369.31723.79313.1713.7467.91825.0065.7814.02213.31926.35322.3915.2786.52026.7568.41017.29117.92127.9808.41118.00171.42230.7235.3。
[0012]
进一步地,所述晶型
ɑ
具有基本上如图4所示的xrpd图谱。
[0013]
进一步地,利用差式扫描量热法,所述晶型
ɑ
在229~234℃的温度下处具有特征吸收峰,优选的,所述晶型
ɑ
具有基本上如图5所示的dsc曲线。
[0014]
进一步地,晶型
ɑ
的红外光谱在3376cm-1
、3084cm-1
、2924cm-1
、2870cm-1
、2833cm-1
、2807cm-1
、2768cm-1
、1693cm-1
、1615cm-1
、1583cm-1
、1532cm-1
、1509cm-1
、1481cm-1
、1429cm-1
、1402cm-1
、1351cm-1
、1307cm-1
、1286cm-1
、1256cm-1
、1233cm-1
、1211cm-1
、1199cm-1
、1176cm-1
、1134cm-1
、1084cm-1
、1051cm-1
、991cm-1
、975cm-1
、961cm-1
、935m-1
、913cm-1
、898cm-1
、861cm-1
、847cm-1
、817cm-1
、728cm-1
、682cm-1
、621cm-1
、614cm-1
、565cm-1
、530cm-1
处具有特征峰。
[0015]
本发明还提供了一种制备上述任一所述的晶型
ɑ
的方法,包括如下步骤:
[0016]
将喹啉化合物和四氢呋喃混合,升温溶解,得到混合液,将混合液经至少一次先加入不良溶剂后减压蒸除溶剂的步骤析出固体,固液分离,收集固体,得到晶型
ɑ
。
[0017]
进一步地,所述不良溶剂选自甲醇、乙醇、丙酮、苯甲醚、乙酸乙酯、二氯甲烷、1,4-二氧六环中的至少一种;和/或,喹啉化合物的质量与四氢呋喃的体积比为1kg:10-20l;和/或,升温溶解的温度为45-55℃;和/或,减压蒸除溶剂的过程中,先在温度为70-80℃的条件下回流3h,在不少于3h的时间内降温至35-45℃下,35-45℃下保温2-3h,在不少于3h的时间内降温至20-30℃,在20-30℃保温3-4h;和/或,所述固液分离选自离心或者过滤。
[0018]
进一步地,喹啉化合物和四氢呋喃混合的混合液与不良溶剂混合的步骤之前还包括预处理,所述预处理为:将喹啉化合物和四氢呋喃混合的混合液浓缩至原料体积的1/3~2/3;和/或,重复加入不良溶剂和减压蒸除溶剂2~3次。
[0019]
进一步地,式(i)所示的喹啉化合物可以完全采用cn102977014a给出的方法通过步骤a-k合成得到制备得到。工艺流程如下:
[0020][0021]
也可以对式ii所示的喹啉化合物中间体和式i所示的喹啉化合物的合成步骤分别进行优化。
[0022]
本发明提供了式ii所示的喹啉化合物中间体的合成方法,包括如下步骤:(1)取式iv所示化合物、碱性催化剂和氯甲酸苯酯,在nmp环境中接触,反应,得到反应液;
[0023]
(2)将步骤(1)制得的反应液与水合肼混合,反应,即得;
[0024][0025]
作为优选的实施方式,步骤(1)包括,采用nmp溶解式iv所示化合物,形成溶液,取溶液与碱性催化剂和氯甲酸苯酯混合,反应,得到反应液;或者,包括采用nmp溶解式iv所示化合物和碱性催化剂,形成溶液,取溶液和氯甲酸苯酯混合,反应,得到反应液。采用上述两种方式更加有利于反应正向进行,提高反应收率。cn102977014a中采用的合成方法为先制备得到式iii所示化合物,通过式iii所示的反应物与水合肼反应,然而本发明研究发现该方法制得的式ii所示喹啉化合物中间体的收率低下,同时含有较多水合肼残留。研究后得知,原因在于式iii所示化合物与水合肼在二氧六环中为非均相反应,导致式ii所示喹啉化合物中间体的收率低下,反应体系蒸除溶剂后过滤后喹啉化合物中间体残留较多水合肼无法除去。而本发明采用一锅反应,式iii所示反应物无需提纯,不仅操作简单,而且能够提高反应收率,同时能够有效清除式ii所示喹啉化合物中间体的水合肼残留。作为优选的实施方式,所述碱性催化剂选自三乙胺、dmap、碳酸钾和吡啶中的至少一种。作为优选的实施方
式,式iv所示化合物的质量与nmp的体积比为1.0kg:4.0-6.0l。作为优选的实施方式,式iv所示化合物与水合肼的摩尔比为1:6.0-8.0。作为优选的实施方式,步骤(1)的反应温度为20-30℃,时间为至少2h,优选为2-3h。作为优选的实施方式,式iv所示化合物与氯甲酸苯酯的摩尔比为1:1.0-1.2。作为优选的实施方式,步骤(2)的反应温度为20-30℃,时间为至少2h,优选为2-6h。作为优选的实施方式,步骤(1)中,在温度为20-30℃的搅拌条件下溶解,溶解时间为0.5-1.0h。作为优选的实施方式,步骤(2)中,在温度不高于30℃的条件下采用滴加的方式混入氯甲酸苯酯。作为优选的实施方式,还包括对步骤(2)反应之后得到的反应液进行提纯的步骤。更优选地,在提纯步骤中,采用碱性水溶液调节反应液的ph为10-12,析晶,固液分离,收集固体,干燥,制得式ii所示的喹啉化合物中间体纯品。其中,固液分离采用常规方式,例如,过滤或者离心。作为优选的实施方式,在加入碱性水溶液之前还包括采用滴加方式加入水的步骤,滴加时间为3-5h,式iv化合物的质量与水的体积比为1kg:15l。作为优选的实施方式,在搅拌下析晶,析晶过程的温度为0-20℃,时间为6-10h;和/或,所述碱性水溶液选自质量百分数为5-15%的氢氧化钠的水溶液。
[0026]
本发明提供了一种如式i所示的喹啉化合物的合成方法,包括,
[0027]
在nmp中,在有机酸催化下,将2,4-二氟苯甲醛与式ii所示的喹啉化合物中间体接触,反应,即得。
[0028]
作为优选的实施方式,所述有机酸选自乙酸、对甲苯磺酸和乙二酸中的至少一种。作为优选的实施方式,2,4-二氟苯甲醛与式ii所示的喹啉化合物中间体的摩尔比为2.8~3.2:1.0。作为优选的实施方式,式ii所示的喹啉化合物中间体的质量与nmp的体积比为1.0kg:5.0~8.0l。作为优选的实施方式,所述的合成方法具体包括如下步骤:(1)将2,4-二氟苯甲醛、nmp和有机酸混合,得到混合液;(2)将步骤(1)得到的混合液与式ii所示的喹啉化合物中间体混合,反应,制得式i所示的喹啉化合物。作为优选的实施方式,步骤(1)中,在搅拌下混合,混合温度为20-30℃,时间为0.5-1h。作为优选的实施方式,步骤(2)中,在搅拌下反应,反应温度为35-45℃,时间为6-16h。作为优选的实施方式,在反应之后还包括对反应液进行提纯的步骤。作为更优选的实施方式,在提纯步骤中,采用碱性水溶液调节反应液的ph为9-10,析晶,过滤,收集固体,干燥,制得式i所示的喹啉化合物粗品。具体地,在搅拌下析晶,析晶过程的温度为0-30℃,时间为4-6h。作为优选的实施方式,所述碱性水溶液选自质量百分数为5%-15%的碳酸钠的水溶液。
[0029]
作为优选的实施方式,在提纯步骤之后还包括取式i所示的喹啉化合物粗品,加入水混合,固液分离;取固体,加入四氢呋喃,加热溶解;然后加水混合,降温,固液分离,取固体干燥,制得式i所示的喹啉化合物纯品,经检测为晶体结构,记为晶型β。作为优选的实施方式,第一次加入的水的体积与式ii所示的喹啉化合物中间体的质量比为10:1。第一次加入的水的体积与式ii所示的喹啉化合物中间体的质量比为3:1。作为优选的实施方式,四氢呋喃的体积与式ii所示的喹啉化合物中间体的质量比为10:1。作为优选的实施方式,在提纯步骤之后还包括取式i所示的喹啉化合物粗品,加入水混合,离心;取固体,加入四氢呋喃,55-65℃下加热溶解;然后加水在55-65℃下搅拌混合3h,在7h内缓慢降温至20-30℃,过滤,干燥,制得式i所示的喹啉化合物纯品。
[0030]
本发明中,式iv化合物可以通过购买,或者采用现有的方法合成制备,例如采用专利文献cn102977014实施例1中公开的合成方法,通过步骤a-h合成得到。
[0031]
本发明还通过对步骤a-h进行改进,以更加适应工业化大规模生产的需求,例如,步骤a中,现有技术采用向3-甲氧基-4羟基苯乙酮中滴加1-溴-3-氯丙烷的方式,而本发明优选地实施方式采用向1-溴-3-氯丙烷中分次加入3-甲氧基-4羟基苯乙酮(例如平均分三次)。步骤b中,现有技术中采用将反应液倒入冰水中,收集有机层,有机层用饱和碳酸氢钠水溶液洗至中性,无水硫酸钠干燥,蒸干溶剂的方式纯化,而本发明优选地实施方式采用将反应液倒入冰水中,然后水相用ch2cl2萃取两次,合并有机相用水洗至水相近中性,减压蒸除溶剂。步骤f中,现有技术采用将反应液加入到冰水的方式淬灭反应。而本发明优选地实施方式采用向反应体系倒入冰水淬灭。通过上述对各步骤综合改进,使得本发明改进后的合成方法适用于大规模生产。
[0032]
本发明还提供了上述任一所述的晶型
ɑ
或者上述任一所述的制备方法制得的晶型
ɑ
在制备用于预防或治疗与c-met激酶抑制剂有关的疾病的药物中的用途;或者,
[0033]
在制备预防和/或治疗肺癌、肝癌、胃癌、结直肠癌、乳腺癌、恶性胶质母细胞瘤、膀胱癌、前列腺癌、卵巢癌或者食管癌的药物中的应用。
[0034]
本发明技术方案,具有如下优点:
[0035]
1、本发明提供的喹啉化合物的晶型
ɑ
,纯度高,在水、胃肠模拟液及磷酸盐缓冲溶液中具有较好的溶解性,利于成药。
[0036]
2、本发明提供的喹啉化合物的晶型
ɑ
,在高温和/或高湿环境中均具有良好的稳定性,而且该晶型具有明显提高的生物利用度,体内吸收更好。
[0037]
3、本发明提供的晶型
ɑ
的制备方法,发明人前期研究考察多种方法后发现通过将喹啉化合物和四氢呋喃混合,升温溶解,经至少一次先加入不良溶剂后减压蒸除溶剂的步骤析出固体,固液分离,收集固体,能够制得晶型
ɑ
,制备方法简单方便,适合大生产。尤其是通过控制将喹啉化合物和四氢呋喃混合的溶液浓缩至原料体积的1/3~2/3;然后重复先加入不良溶剂后减压蒸除溶剂2~3次;相比于直接加入同比例的不良溶剂来说,能够明显提高晶型
ɑ
的产率。
[0038]
4、本发明的喹啉化合物通过如下方法制备,取2,4-二氟苯甲醛与式ii所示的喹啉化合物中间体反应后的反应液,采用碱性水溶液调节反应液的ph为10-12,析晶,固液分离,收集固体,干燥,即得喹啉化合物粗品,该方法能够有效清除未反应的原料和其他可能的未知杂质(将水合肼和其他水溶性杂质留在碱性水溶液中),提高喹啉化合物的纯度。发明人前期考察了多种溶剂,例如乙醇、异丙醇、thf等大量溶剂,均无法实现均相反应,意外发现采用nmp为反应溶剂将2,4-二氟苯甲醛与式ii所示的喹啉化合物中间体接触,反应,不仅可以实现均相反应,明显提高喹啉化合物的产率,而且也大大提高了粗品纯度,降低了纯化难度,操作方便,有利于工业规模化生产,粗品收率高达95%以上,纯度高达94%以上,经纯化后符合中国药典及ich相关指导原则中原料药质量要求。此外,采用nmp为反应溶剂还极大降低了2,4-二氟苯甲醛的用量降低至2.8-3.2摩尔当量,即只需现有技术中2,4-二氟苯甲醛用量的50%左右,即2,4-二氟苯甲醛与式ii所示的喹啉化合物中间体的摩尔比为2.8-3.2:1.0,有利于环保和生产安全性。
[0039]
进一步地,通过控制2,4-二氟苯甲醛与式ii所示的喹啉化合物中间体2.8-3.2:1.0,可以高效(收率>95%,纯度>94%)生产出喹啉化合物粗品,合成方法的后处理简便,使用碳酸钠或氢氧化钠水溶液调节ph10-12后析晶洗涤即的喹啉化合物粗品。
[0040]
通过式ii所示的喹啉化合物中间体的质量与nmp的体积比为1.0kg:5.0-8.0l,该质量体积比下,能够充分溶解式ii所示的喹啉化合物中间体和2,4-二氟苯甲醛,反应均相快速,副产物少,进而使式ii所示的喹啉化合物中间体充分转化为喹啉化合物。
[0041]
5、本发明的喹啉化合物的合成方法,在式ii所示的喹啉化合物中间体的合成中,发明人前期考察了多种溶剂,例如dcm、thf、二氧六环、dmso、乙酸乙酯等,无法实现两步均为均相反应,意外发现采用nmp为反应溶剂,将式iv化合物、碱性催化剂和氯甲酸苯酯在nmp接触下反应,然后与水合肼混合反应的合成方法,不仅可以实现两步均相反应,大大提高如式ii所示喹啉化合物中间体的产率,而且仅需要最后一步提纯工艺,中间无需蒸除原反应溶剂后更换新溶剂,操作方便,有利于工业规模化生产,此外,采用nmp为溶剂实现均相反应,还极大降低了水合肼的用量(由60.0eq降低至6.0~8.0eq,即本发明只需要采用现有技术水合肼用量的0.1-0.13倍),有利于环保和生产安全性。
[0042]
此外,本发明既可以先采用nmp溶解式iv所示化合物,形成溶液,取溶液与碱性催化剂和氯甲酸苯酯混合,反应,得到反应液;也可以采用nmp溶解式iv所示化合物和碱性催化剂,形成溶液,取溶液和氯甲酸苯酯混合,反应,得到反应液。上述两种方式均可以实现反应液与水合肼的均相反应。
附图说明
[0043]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0044]
图1是实施例1中式(i)所示的喹啉化合物晶型β的x射线衍射图;
[0045]
图2是实施例2中式(i)所示的喹啉化合物晶型β的x射线衍射图;
[0046]
图3是实施例2中式(i)所示的喹啉化合物晶型β的dsc曲线;
[0047]
图4是实施例3中式(i)所示的喹啉化合物晶型
ɑ
的x射线衍射图;
[0048]
图5是实施例3中式(i)所示的喹啉化合物晶型
ɑ
的dsc曲线;
[0049]
图6是实施例3中式(i)所示的喹啉化合物晶型
ɑ
的ir图;
[0050]
图7是实施例4中式(i)所示的喹啉化合物晶型
ɑ
的xrpd图谱;
[0051]
图8是实施例5中式(i)所示的喹啉化合物晶型
ɑ
的xrpd图谱;
[0052]
图9是实验例1中实施例3制备的晶型
ɑ
灌胃给药的平均血药浓度-时间曲线;
[0053]
图10是实验例1中实施例2制备的晶型β灌胃给药的平均血药浓度-时间曲线;
[0054]
图11是实验例2中晶型
ɑ
的高湿稳定性xrpd对比图;
[0055]
图12是实验例2中晶型
ɑ
的高温稳定性xrpd对比图;
[0056]
图13是实验例2中晶型β的高湿稳定性xrpd对比图;
[0057]
图14是实验例2中晶型β的高温稳定性xrpd对比图;
[0058]
图15是实验例3中晶型
ɑ
在fassif-v1中混悬24h的xrpd图谱;
[0059]
图16是实验例3中晶型
ɑ
在ph 6.8磷酸盐缓冲液中混悬24h的xrpd图谱;
[0060]
图17是实验例3中晶型
ɑ
在纯水中混悬24h的xrpd图谱;
[0061]
图18是实施例2中式(i)所示的喹啉化合物晶型β的1h-nmr图谱。
具体实施方式
[0062]
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
[0063]
本技术采用的实验仪器及测试条件具体如下:
[0064]
x射线粉末衍射仪xrpd公司/型号:bruker d8 advance;方法:检测器:lynxeye_xe_t(1d mode),开启角度:2.94
°
,扫描模式:continuous psd fast,辐射:cu/k-alpha1x-射线源功率:40kv,40ma,步长:0.02
°
,每步时间:0.12s/步,扫描范围:3
°
to 40
°
,主光束路径裂缝:双主电动狭缝10.0mm,索拉狭缝2.5
°
,次光束路径裂缝:探测器索拉狭缝2.5
°
双二级电动狭缝5.2mm,样品旋转速度:15rpm。
[0065]
差式量热扫描仪dsc公司/型号:ta discovery 2500或q2000;方法:样品盘:tzero盘andtzero hermetic扎孔盖;温度范围:30至300℃;升温速率:10℃/min;氮流速:50ml/min;样品重量:1-2mg。
[0066]
红外分光光度计ir,公司/型号:nicolet 6700,thermo scientific,方法:样品扫描次数32次,背景扫描次数:32次,分辨率4,波长范围4000to 525cm-1,基线矫正是,光速0.4747,光圈150,窗口钻石。
[0067]
本发明实验例2-5中式i所示的喹啉化合物的纯度通过高效液相法检测,取式i所示的喹啉化合物10mg加甲醇10ml制得供试品溶液,取供试品溶液注入液相中检测,色谱条件如下:
[0068][0069]
式ii所示的喹啉化合物中间体的纯度通过高效液相法检测,取式ii所示的喹啉化合物10mg加甲醇10ml制得供试品溶液,取供试品溶液注入液相中检测,色谱条件如下:
[0070]
[0071][0072]
式iii所示的喹啉化合物中间体的纯度通过高效液相法检测,取式iii所示的喹啉化合物10mg加甲醇10ml制得供试品溶液,取供试品溶液注入液相中检测,色谱条件如下:
[0073][0074]
中间体iv的纯度通过高效液相法检测,取中间体iv10mg加甲醇10ml制得供试品溶液,取供试品溶液注入液相中检测,色谱条件如下:
[0075][0076]
中间体v、vi、vii、viii、ix和xi的纯度通过高效液相法检测,取中间体10mg加甲醇10ml制得供试品溶液,取供试品溶液注入液相中检测,色谱条件如下:
[0077]
[0078][0079]
实施例1晶型β的制备
[0080]
按照cn102977014a实施例1的方法制备式(i)所示的喹啉化合物,测定获得的式(i)所示的喹啉化合物的xrpd图谱如图1所示,其为晶体,记为晶型β,特征峰见表1所示。
[0081]
表1晶型β的特征峰
[0082][0083]
实施例2晶型β的制备
[0084]
本实施例提供了一种晶型β的制备方法,其流程路线如下,
[0085][0086]
具体的制备方法包括如下步骤:
[0087]
(1)步骤a:中间体xi的合成
[0088]
将1-溴-3-氯丙烷91.5kg和无水碳酸钾80.3kg,加入到反应釜中,加入330kgdmf,然后室温加入3-甲氧基-4羟基苯乙酮23.0kg,室温反应3.5h,然后再室温加入3-甲氧基-4羟基苯乙酮23.0kg,室温反应5h,最后室温加入3-甲氧基-4羟基苯乙酮23.0kg,室温反应15.5h,将所得反应液倒入690kg冰水中,搅拌2.0h,过滤干燥得92.0kg固体(中间体xi),收率92.0%,纯度99.1%。
[0089]
(2)步骤b和c:中间体ix的合成
[0090]
将dcm740kg,步骤a所得中间体92.0kg加入到反应釜中并冷却至-15~-10℃,然后用蠕动泵将发烟硝酸(89.6kg,3.75eq)缓慢加入到反应釜中,-15~-10℃下反应2.0h,将体系倒入冰水中,然后水相用ch2cl2萃取两次,合并有机相用水洗至水相近中性,减压蒸除溶剂。加入n,n-二甲基甲酰胺二甲缩醛(dmf-dma)153kg和甲苯397kg,在温度为95℃条件下反应16h减压蒸除溶剂,再加入甲基叔丁基醚(mtbe)136kg,降温至-15~-10℃并搅拌3h,过滤干燥得85.0kg固体(中间体ix),收率65.0%,纯度95.3%。
[0091]
(3)步骤d:中间体viii的合成
[0092]
将乙酸710kg抽入2000l反应釜中,将步骤b和c所得中间体(85.0kg,1.0eq)加入釜中并升温至70℃。将fe(总质量69.2kg)分批加入并控温为95~100℃。反应体系于95~100℃反应40min。过滤收集滤液,滤液在搅拌下加入mtbe1700l(20v),在-5~0℃下搅拌0.5h,过滤得滤饼并用水680l(8v)清洗,滤饼在50℃减压干燥,得34.8kg产物(中间体viii),产率52.5%,纯度97.0%。
[0093]
(4)步骤e:中间体vii的合成
[0094]
将乙腈(ch3cn)174l抽入釜中,将步骤d所得中间体34.7kg及4-甲基哌啶51.4kg加入反应釜中,75~80℃反应4h。减压蒸除溶剂,向浓缩体系中加入乙酸乙酯(etoac)174l,搅拌析晶3.5h,过滤收集滤饼。滤饼在50℃减压干燥,得产物(中间体vii)35.8kg,产率83.5%。
[0095]
(5)步骤f:中间体vi的合成
[0096]
将ch3cn107l及氧氯化磷5.5kg(pocl3,6.9eq)抽入反应釜,将步骤e所得中间体35.8kg加入反应釜,75-80℃反应4h。45℃减压蒸除溶剂,向反应体系倒入冰水573l淬灭。用50%(w/v)的氢氧化钠水溶液调节ph为11~12。加40~45℃水573l稀释至盐溶解,将料液转移至离心机甩滤,用水14.3l洗涤滤饼5次,滤饼在50℃减压干燥得产物31.5kg(中间体vi),产率83.6%,纯度98.4%。
[0097]
(6)步骤g:中间体v的合成
[0098]
将乙醇100l抽入到反应釜,加热回流30min后,放出,将氯苯100l抽入到反应釜,洗涤30min后,放出。将氯苯157l抽入反应釜,将步骤f所得中间体31.4kg及2-氟-4硝基苯酚21kg加入反应釜,135~140℃反应12h。反应体系降温至-10~-5℃析晶2h。过滤得到滤饼,用dcm 628l溶解滤饼,用10%(w/w)k2co3溶液502l分5次清洗有机相至水层近无色。用5%(w/w)食盐水洗涤有机相两次,有机相35~40℃浓缩,加入异丙醚251l20~30℃析晶4h,抽滤得到滤饼。滤饼用etoh314l溶解并加热至80℃搅拌0.5h,降温至20~30℃析晶5h,抽滤得到滤饼,滤饼用少量etoh9.4l清洗,干燥得产物21.0kg(中间体v),产率50.0%,纯度97.7%。
[0099]
(7)步骤h:中间体iv的合成
[0100]
将乙醇100l抽入到反应釜,加热回流30min后,放出(重复2次)。将乙醇252l抽入反应釜。将浓hcl加入反应釜,升温至60~65℃并将fe 15kg(6eq.)加入反应釜搅拌10min。分批加入步骤g所得中间体21.0kg并升温至75~80℃反应2h。降温至50~55℃加入活性炭的etoh42l溶液升温至75~80℃反应0.5h,加入水42l75~80℃回流10min,过滤得到滤液。冷却至10~15℃并用5%(w/w)k2co3调节ph为10~11。加1260l水稀释并在15~20℃析晶2h。过滤得到滤饼并用水清洗至滤液ph至约7,滤饼在45℃减压干燥得产物(即为式iv化合物)15.8kg,产率80.0%,纯度99.0%。
[0101]
(8)步骤i和步骤j:式ii所示的喹啉化合物中间体的合成
[0102]
将式iv化合物15.8kg(36mol,1.0eq)和nmp79l(5v)加入反应釜中,控制反应釜内温度为20~30℃,搅拌0.75h至反应液溶清,加入吡啶5.7kg(72mol,2.0eq)到反应釜中。控制反应釜内温度为20~30℃,缓慢滴加氯甲酸苯酯6.2kg(39.6mol,1.1eq)到反应釜中,20~30℃搅拌反应2.5h,采用hplc检查式iv化合物反应完全(<0.5%)及式iii化合物的纯度(>95%),加入80%(w/w)水合肼水溶液15.7kg(252mol,7.0eq)到反应釜中,20~30℃搅拌反应3h。缓慢滴加纯化水237l(15v)到反应釜中(滴加时间4.0h),控温20~30℃缓慢滴加10%(w/w)naoh水溶液调节反应液ph为10~11。调节反应釜内温0~20℃,搅拌析晶8h。过滤,使用纯化水洗涤滤饼。滤饼45℃真空干燥20h,得产物(式ii所示的喹啉化合物中间体)11.8kg,产率95.2%,纯度95.7%。
[0103]
(9)步骤k:式i所示的喹啉化合物的合成与提纯
[0104]
向反应釜加入2,4-二氟苯甲醛9.9kg(3.0eq)、nmp58.5l(5v)及acoh0.4kg(0.3eq),20-30℃搅拌0.5小时,加入式ii所示的喹啉化合物中间体11.7kg(1.0eq)至反应釜。调节温度35~45℃,搅拌反应8h。调节温度20~30℃,缓慢滴加na2co3溶液调节反应液ph约9~10,20~30摄氏度搅拌析晶5h。过滤,用纯化水漂洗滤饼,干燥后得粗品14.0kg,产率95.6%,纯度94.8%。
[0105]
将干燥后的粗品14.0kg返釜,加入纯化水117l(10v),20-30℃打浆6小时,离心,使用纯化水23.4l(2v)漂洗滤饼。将所得滤饼返釜,加入四氢呋喃117l(10v)到反应釜中,升温至55-65℃,保温搅拌溶清后,缓慢滴加纯化水35.1l(3v),55-65℃搅拌3小时,控制约7小时缓慢降温至20~30℃,过滤使用纯化水23.4l(2v)漂洗滤饼。45-55℃干燥20h,得产物10.1kg,产率69.1%,纯度99.3%。xrpd图谱见图2所示,与实施例1中的图1基本一致,为晶型β。
[0106]
进一步的检测晶型β的dsc曲线如图3所示,其中,dsc曲线显示晶型β在90.5℃(peak)及234.0℃(peak)存在吸热峰,90.5℃为脱水吸热峰,234.0℃为熔融吸热峰。检测晶型β的质谱和核磁,核磁图谱如图18,确认晶型β的结构式如式(i)所示。
[0107]
esi-ms[m h]622.2
[0108]1h-nmr
[0109][0110]
表2qbh-1961h-nmr数据及归属
[0111][0112]
实施例3晶型
ɑ
的制备
[0113]
本实施例提供了一种晶型
ɑ
及其制备方法,其制备方法包括如下步骤:
[0114]
称取实施例2的晶型β10.1kg及thf 200l到反应釜,反应釜温度调至45~55℃,搅拌溶清。过滤,收集滤液,将滤液真空浓缩至50l,然后投入无水乙醇50l,再次真空浓缩至50l,同操作添加无水乙醇并浓缩置换的操作重复三次。然后投入无水乙醇50l到反应釜,调节反应釜内温至70~80℃,搅拌回流3h。3小时内缓慢降温至35~45℃,保温2.5h。3小时内缓慢降温至20~30℃,搅拌3.5h。过滤,无水乙醇10l漂洗滤饼,取样检测固体xrpd,图4所示,与晶型β属于不同晶体,记为晶型
ɑ
。然后在45~55℃真空干燥15h。得产物8.8kg,产率87%,纯度99.6%。
[0115]
表3晶型
ɑ
的特征峰
[0116][0117]
进一步的检测晶型
ɑ
的dsc曲线和ir图谱如图5和6所示,其中,dsc曲线显示晶型
ɑ
在233.4℃存在一吸收峰。ir图谱显示在3376cm-1
、3084cm-1
、2924cm-1
、2870cm-1
、2833cm-1
、2807cm-1
、2768cm-1
、1693cm-1
、1615cm-1
、1583cm-1
、1532cm-1
、1509cm-1
、1481cm-1
、1429cm-1
、1402cm-1
、1351cm-1
、1307cm-1
、1286cm-1
、1256cm-1
、1233cm-1
、1211cm-1
、1199cm-1
、1176cm-1
、1134cm-1
、1084cm-1
、1051cm-1
、991cm-1
、975cm-1
、961cm-1
、935m-1
、913cm-1
、898cm-1
、861cm-1
、847cm-1
、817cm-1
、728cm-1
、682cm-1
、621cm-1
、614cm-1
、565cm-1
、530cm-1
处具有特征峰。
[0118]
实施例4晶型
ɑ
的制备
[0119]
本实施例提供了一种晶型
ɑ
及其制备方法,其制备方法包括如下步骤:称取实施例2的晶型β10.0g及thf 200ml到圆底烧瓶,反应釜温度调至45~55℃,搅拌溶清。过滤,收集滤液,将滤液投入无水乙醇50ml,真空浓缩至50ml,按照上述操作添加无水乙醇并浓缩置换的操作重复三次。然后投入无水乙醇50ml到圆底烧瓶,调节反应釜内温至70~80℃,搅拌回流4h。3小时内缓慢降温至20~30℃,搅拌5h。过滤,乙醇10ml漂洗滤饼,然后在45~55℃真空干燥15h。得产物5.1g,产率51%,纯度99.7%。xrpd图谱见图7所示,与实施例3中的图4基本一致。
[0120]
实施例5晶型
ɑ
的制备
[0121]
本实施例提供了一种晶型
ɑ
及其制备方法,其制备方法包括如下步骤:称取实施例2的晶型β10.0g及thf(200ml)到圆底烧瓶,反应釜温度调至45~55℃,搅拌溶清。过滤,收集滤液,将滤液真空浓缩至50ml,然后投入无水二氧六环50ml,再次真空浓缩至50ml,添加无水二氧六环并浓缩置换的操作重复三次。然后投入无水二氧六环50ml到圆底烧瓶,调节反应釜内温至70~80℃,搅拌回流3h。3小时内缓慢降温至35~45℃,保温2.5h。3小时内缓慢降温至20~30℃,搅拌3.5h。过滤,二氧六环10ml漂洗滤饼,然后在45~55℃真空干燥15h。得产物7.8g,产率78%,纯度99.6%。xrpd图谱见图8所示,与实施例3中的图4基本一致。
[0122]
实验例1药代动力学实验
[0123]
分别将实施例2制备的喹啉化合物晶型β和实施例3制备的喹啉化合物晶型
ɑ
作为受试物,制成含喹啉化合物的cmc水溶液混悬液(cmc质量分数为0.5%,喹啉化合物浓度为3mg/ml)。
[0124]
8只wistar大鼠,雄性,随机分为两组,每组4只,以35mg/kg动物体重的剂量分别灌
胃给予上述两种受试药物溶液,取血方式:眼眶取血,取血时间点:0.25h、0.5h、1h、2h、3h、4h、6h、8h、12h、18h、24h、32h、48h、72h分别取血;血液处理方法:甲醇沉淀蛋白,涡旋1min后离心(11000rpm,5min),取上清液进液质测定;血液中药物的含量测定方法:液质联用法,测试条件为:仪器型号(agilent 1260s,四级杆自动进样,dad检测器),色谱柱(固定相c18,型号sb-c18),流速(1.0μl/min),进样量(20μl),流动相(磷酸盐缓冲液,25mm磷酸二氢钠,磷酸调节ph=2.5),梯度洗脱,归一化法分析。
[0125]
表4晶型
ɑ
的药动参数(n=4)
[0126][0127]
表5晶型β的药动参数(n=4)
[0128][0129]
由图9-10和上表结果可知,晶型
ɑ
生物利用度明显高于晶型β,体内吸收明显优于晶型β。
[0130]
实验例2稳定性
[0131]
分别将实施例2制备的晶型β和实施例3制备的晶型
ɑ
作为受试物进行如下检测。分别将受试物置于60℃下的高温条件下以及25℃,湿度92.5%rh的高湿条件下测定上述两种晶型的稳定性,分别在实验前以及两个实验条件下放置的第5天和第10天同一时刻取样,观察外观,采用hplc峰面积归一法测定纯度和x-射线粉末衍射,见表6-7和图11-14所示。
[0132]
表6晶型
ɑ
的稳定性实验结果
[0133][0134]
表7晶型β的稳定性实验结果
[0135][0136][0137]
结果显示,两个批次的晶型
ɑ
均具有高温稳定性和高湿稳定性,而晶型β两批产物在高温10天x射线粉末衍射花纹有变化,均出现新的衍射峰(约25.10
°
),说明晶型β高温稳定性差。
[0138]
实验例3
[0139]
称取20mg优势晶型α,分别加入10mlfassif-v1(饥饿状态下模拟肠液)、ph6.8磷酸
盐缓冲液和水,在37℃400rpm转速下混悬24h。将混悬液在37℃14000rpm条件下离心,将离心所得上清液用hplc进行溶解度检测,测试剩余上清液ph,将离心所得固体沉淀用xrpd进行检测。
[0140]
在fassif-v1、ph 6.8磷酸盐缓冲液及纯水中无晶型转变,见图15-17所示。
[0141]
实验例4式i所示的喹啉化合物的合成与纯化方法的考察
[0142]
分别采用下述四种方法制备式i所示的喹啉化合物纯品。
[0143]
方法一:向反应釜加入2,4-二氟苯甲醛9.24kg(2.8eq)、nmp 58.5l(5v)及acoh 0.4kg(0.3eq),20-30℃搅拌1小时,加入按照实施例1的方法制得的式ii所示的喹啉化合物中间体11.7kg(1.0eq)至反应釜。调节温度35-45℃,搅拌反应14h。调节温度20-30℃,缓慢滴加15wt%的na2co3溶液调节反应液ph约9-10,20-30摄氏度搅拌析晶5h。过滤,用纯化水漂洗滤饼,干燥后得粗品14.1kg,产率96.3%,纯度94.5%。
[0144]
将干燥后得粗品14.1kg返釜,加入纯化水117l(10v),20-30℃打浆6小时,离心,使用纯化水23.4l(2v)漂洗滤饼。将所得滤饼返釜,加入四氢呋喃117l(10v)到反应釜中,升温至55-65℃,保温搅拌溶清后,缓慢滴加纯化水35.1l(3v),55-65℃搅拌3小时,控制约7小时缓慢降温至20-30℃,过滤使用纯化水23.4l(2v)漂洗滤饼。45-55℃干燥20h,得产物10.4kg,产率71.1%,纯度99.4%。
[0145]
方法二:向反应釜加入2,4-二氟苯甲醛10.6kg(3.2eq)、nmp58.5l(5v)及acoh 0.4kg(0.3eq),20-30℃搅拌1小时,加入按照实施例1的方法制得的式ii所示的喹啉化合物中间体11.7kg(1.0eq)至反应釜。调节温度35-45℃,搅拌反应16h。调节温度20-30℃,缓慢滴加质量百分数为8%的na2co3溶液调节反应液ph约9-10,20-30摄氏度搅拌析晶6h。过滤,用纯化水漂洗滤饼,干燥后得粗品14.2kg,产率97.0%,纯度94.1%。
[0146]
将干燥后得粗品14.2kg,加入纯化水117l(10v),20-30℃打浆6小时,离心,使用纯化水23.4l(2v)漂洗滤饼。将所得滤饼返釜,加入四氢呋喃117l(10v)到反应釜中,升温至55-65℃,保温搅拌溶清后,缓慢滴加纯化水35.1l(3v),55-65℃搅拌3小时,控制约7小时缓慢降温至20-30℃,过滤使用纯化水23.4l(2v)漂洗滤饼。45-55℃干燥20h,得产物10.0kg,产率68.4%,纯度99.4%。
[0147]
方法三:基本采用实施例2的合成方法,与实施例2的区别仅在于在喹啉化合物的合成步骤中,采用同体积的乙醇替代nmp。
[0148]
具体步骤为:向反应釜加入2,4-二氟苯甲醛9.9kg(3.0eq)、乙醇58.5l(5v)及acoh0.4kg(0.3eq),20-30℃搅拌0.5小时,加入按照实施例1的方法制得的式ii所示的喹啉化合物中间体11.7kg(1.0eq)至反应釜。调节温度35-45℃,搅拌反应8h。调节温度20-30℃,缓慢滴加na2co3溶液调节反应液ph约9-10,20-30摄氏度搅拌析晶5h。过滤,用纯化水漂洗滤饼,干燥后得粗品9.9kg,产率67.8%,纯度85.2%。
[0149]
方法四:基本采用实施例2的合成方法,与实施例2的区别仅在于在喹啉化合物的合成步骤中,采用同体积的异丙醇替代nmp。
[0150]
具体步骤为:向反应釜加入2,4-二氟苯甲醛9.9kg(3.0eq)、异丙醇58.5l(5v)及acoh 0.4kg(0.3eq),20-30℃搅拌0.5小时,加入按照实施例1的方法制得的式ii所示的喹啉化合物中间体11.7kg(1.0eq)至反应釜。调节温度35-45℃,搅拌反应8h。调节温度20-30℃,缓慢滴加na2co3溶液调节反应液ph约9-10,20-30摄氏度搅拌析晶5h。过滤,用纯化水漂
洗滤饼,干燥后得粗品9.7kg,产率66.4%,纯度84.1%。
[0151]
实验例5式ii所示的喹啉化合物中间体合成方法的考察
[0152]
分别采用下述三种方法制备式ii所示的喹啉化合物中间体。
[0153]
方法一:将式iv化合物15.8kg(36mol,1.0eq)和nmp 79l(5v)加入反应釜中,控制反应釜内温度为20~30℃,搅拌1h至反应液溶清,加入吡啶5.7kg(72mol,2.0eq)到反应釜中。控制反应釜内温度为20~30℃,缓慢滴加氯甲酸苯酯6.2kg(39.6mol,1.1eq)到反应釜中,20~30℃搅拌反应2h,采用hplc检查式iv化合物反应完全(<0.5%)及式iii化合物的纯度(>95%),加入80%(w/w)水合肼水溶液15.7kg(252mol,7.0eq)到反应釜中,20~30℃搅拌反应2h。缓慢滴加纯化水237l(15v)到反应釜中(滴加时间4.0h),控温20~30℃缓慢滴加10%(w/w)naoh水溶液调节反应液ph为10~11。调节反应釜内温0~20℃,搅拌析晶6h。过滤,使用纯化水洗涤滤饼。滤饼45℃真空干燥20h,得产物(式ii所示的喹啉化合物中间体)12.0kg,产率97.0%,纯度95.1%。
[0154]
方法二:将式iv化合物15.8kg(36mol,1.0eq)和nmp 79l(5v)加入反应釜中,控制反应釜内温度为20~30℃,搅拌1h至反应液溶清,加入吡啶5.7kg(72mol,2.0eq)到反应釜中。控制反应釜内温度为20~30℃,缓慢滴加氯甲酸苯酯6.2kg(39.6mol,1.1eq)到反应釜中,20~30℃搅拌反应6h,采用hplc检查式iv化合物反应完全(<0.5%)及式iii化合物的纯度(>95%),加入80%(w/w)水合肼水溶液15.7kg(252mol,7.0eq)到反应釜中,20~30℃搅拌反应6h。缓慢滴加纯化水237l(15v)到反应釜中(滴加时间4.0h),控温20~30℃缓慢滴加10%(w/w)naoh水溶液调节反应液ph为10~11。调节反应釜内温0~20℃,搅拌析晶10h。过滤,使用纯化水洗涤滤饼。滤饼45℃真空干燥20h,得产物(式ii所示的喹啉化合物中间体)11.78kg,产率95.2%,纯度95.6%。
[0155]
方法三:基本采用实施例2的合成方法,与实施例2的区别仅在于合成规模等比降低,采用相应体积二氯甲烷替代nmp。
[0156]
具体步骤为:将式iv化合物15.8g(36mmol,1.0eq)和dcm 79ml(5v)加入反应釜中,控制反应釜内温度为20~30℃,搅拌0.75h至反应液溶清,加入吡啶5.7g(72mmol,2.0eq)到反应釜中。控制反应釜内温度为20~30℃,缓慢滴加氯甲酸苯酯6.2g(39.6mmol,1.1eq)到反应釜中,20~30℃搅拌反应2.5h,采用hplc检查式iv化合物反应不完全(>30%)及式iii化合物的纯度(<60%),hplc检测结果提示当前反应液大量式iv化合物剩余,不适合进行后续反应,故而未进行后续反应。
[0157]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。