1.本发明涉及新型化合物及其用于治疗自身免疫性疾病的用途。
背景技术:
2.可以通过免疫应答保护人体免受病原体的侵害。通常将针对例如病毒和细菌等外来微生物的生物防御机制分为先天性免疫(innate immunity)和特异性免疫(specific immunity),其由主要由免疫相关细胞分泌的细胞因子介导。
3.免疫系统用于保护身体免受抗原(antigen)(即,有害的外来物质)的侵害。这些抗原的类型包括来自其它人类或动物的细菌、病毒、毒素、癌细胞以及血液和组织。免疫系统产生抗体来破坏这些有害物质。如果存在自身免疫性病症,则免疫系统无法区分其身体器官和有害抗原,并且可能会破坏正常组织。通过如上所述的此类应答诱发的疾病为自身免疫性疾病(autoimmune disease)。
4.芳烃受体(aryl hydrocarbon receptor,ahr)为属于per-arnt-sim(pas)超家族的配体依赖性转录因子,并且主要在屏障组织的免疫细胞、上皮细胞、内皮细胞和基质(stromal)细胞中表达。ahr为环境感受器(environmental sensor)并且不仅检测例如环境污染物(例如,二噁英)等生物异源性(xenobiotic)配体还检测由细胞、微生物和食品产生的生理性配体。
5.ahr的失活形式在细胞质中与hsp90:xap2:p23:src伴侣形成复合物(ahr伴侣复合物),并且保持对配体具有高亲和力的结构。当ahr在配体结合之后被激活时,复合物移动至细胞核,并且ahr从伴侣复合物中分离并且与位于靶基因的上游调节区的ahr响应性dna元件(生物异源物质响应元件(xenobiotic response element,xre))结合以调节靶基因的表达。可以开发可以在体内激活ahr的无毒的免疫调节配体作为新的用于自身免疫性疾病的治疗剂。
技术实现要素:
6.发明要解决的问题
7.本发明的目的在于提供新型化合物、其立体异构体或药学上可接受的盐。
8.此外,本发明的另一目的在于提供对于预防和治疗自身免疫性疾病有用的新型化合物、其立体异构体或药学上可接受的盐。
9.此外,本发明的另一目的在于提供用于预防或治疗自身免疫性疾病的药物组合物,其包含新型化合物、其立体异构体或药学上可接受的盐。
10.用于解决问题的方案
11.1.一种由下式1表示的化合物、其立体异构体或药学上可接受的盐:
12.[式1]
[0013][0014]
(其中r1至r4各自独立地为氢或卤素,r5和r6各自独立地为氢或c
1-c5烷基,
[0015]
a为c
5-c
12
的单环状基团或双环状基团,
[0016]
环状基团的各环由1至3个杂原子取代,并且
[0017]
环状基团由卤素、c
1-c5烷基或c
1-c5烷氧基取代)。
[0018]
2.根据上述1的化合物、其立体异构体或药学上可接受的盐,其中a选自由以下环状基团组成的组:
[0019][0020][0021]
(其中q1至q
15
各自独立地为c、n或s,并且r7至r
30
各自独立地为氢、卤素、c
1-c3烷基或c
1-c3烷氧基,并且如果q4为n,则r
11
不存在)。
[0022]
3.根据上述1的化合物、其立体异构体或药学上可接受的盐,其中a选自由以下环状基团组成的组:
[0023][0024]
(其中r7至r
30
各自独立地为氢、卤素、c
1-c3烷基或c
1-c3烷氧基)。
[0025]
4.根据上述1的化合物、其立体异构体或药学上可接受的盐,其中a选自由以下环状基团组成的组:
[0026][0027]
(其中r7至r
24
各自独立地为氢、卤素、c
1-c3烷基或c
1-c3烷氧基)。
[0028]
5.根据上述1的化合物、其立体异构体或药学上可接受的盐,其中a选自由以下环状基团组成的组:
[0029][0030]
(其中r9至r
16
各自独立地为氢、卤素、c
1-c3烷基或c
1-c3烷氧基)。
[0031]
6.根据上述1的化合物、其立体异构体或药学上可接受的盐,其中r2和r3各自独立
地为f、cl或br。
[0032]
7.根据上述1的化合物、其立体异构体或药学上可接受的盐,其中所述化合物选自由以下化合物组成的组:
[0033]
n-(5-溴-6-甲基吡啶-2-基)-2-(1-甲基-1h-吲哚-3-基)乙酰胺,
[0034]
n-(5-溴-6-甲基吡啶-2-基)-2-(1h-吲哚-3-基)乙酰胺;
[0035]
n-(5-溴-6-甲基吡啶-2-基)-2-(5-氯-1h-吲哚-3-基)乙酰胺;
[0036]
n-(苯并[二]噻唑-2-基)-2-(1h-吲哚-3-基)乙酰胺;
[0037]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1h-吲哚-3-基)乙酰胺;
[0038]
n-(5-氯-6-氟吡啶-2-基)-2-(1h-吲哚-3-基)乙酰胺;
[0039]
2-(5-氯-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺;
[0040]
2-(1h-吲哚-3-基)-n-(3,4,5-三甲氧基苯基)乙酰胺;
[0041]
2-(5-氯-1h-吲哚-3-基)-n-(3,4,5-三甲氧基苯基)乙酰胺;
[0042]
n-(3,5-二氯苯基)-2-(1h-吲哚-3-基)乙酰胺;
[0043]
2-(5-氯-1h-吲哚-3-基)-n-(3,5-二氯苯基)乙酰胺;
[0044]
n-(5-溴-6-甲基吡啶-2-基)-2-(5-氟-1h-吲哚-3-基)乙酰胺;
[0045]
2-(5-氯-1h-吲哚-3-基)-n-(吡啶-4-基)乙酰胺;
[0046]
n-(苯并[二]噻唑-2-基)-n-甲基-2-(1-甲基-1h-吲哚-3-基)乙酰胺;
[0047]
n-(苯并[二]噻唑-2-基)-2-(1h-吲哚-3-基)-n-甲基乙酰胺;
[0048]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1h-吲哚-3-基)-n-甲基乙酰胺;
[0049]
n-(5-氯-6-氟吡啶-2-基)-2-(1h-吲哚-3-基)-n-甲基乙酰胺;
[0050]
2-(5-氯-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)-n-甲基乙酰胺;
[0051]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1-甲基-1h-吲哚-3-基)-n-甲基乙酰胺;
[0052]
n-(5-氯-6-氟吡啶-2-基)-n-甲基-2-(1-甲基-1h-吲哚-3-基)乙酰胺;
[0053]
2-(5-氯-1-甲基-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)-n-甲基乙酰胺;
[0054]
2-(5-氯-1-甲基-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺;
[0055]
n-(苯并[二]噻唑-2-基)-2-(1-甲基-1h-吲哚-3-基)乙酰胺;
[0056]
n-(苯并[二]噻唑-2-基)-2-(5-氟-1h-吲哚-3-基)-n-甲基乙酰胺;
[0057]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1-甲基-1h-吲哚-3-基)乙酰胺;
[0058]
n-(苯并[二]噻唑-2-基)-2-(5-氟-1h-吲哚-3-基)乙酰胺;
[0059]
n-(苯并[二]噻唑-2-基)-2-(6-氯-1h-吲哚-3-基)乙酰胺;
[0060]
2-(5-氯-1h-吲哚-3-基)-n-(噻唑-2-基)乙酰胺;
[0061]
2-(5-氯-1h-吲哚-3-基)-n-(喹啉-2-基)乙酰胺;和
[0062]
2-(5-氯-1h-吲哚-3-基)-n-(4,5,6,7-四氢苯并[二]噻唑-2-基)乙酰胺。
[0063]
8.一种药物组合物,其包含根据上述1至7中任一项的化合物、其立体异构体或药学上可接受的盐。
[0064]
9.根据上述8的药物组合物,其中所述组合物用于治疗或预防自身免疫性疾病。
[0065]
10.根据上述8的药物组合物,其中自身免疫性疾病为选自由多发性硬化、炎症性肠病、移植物抗宿主病、哮喘、特应症(atopy)、银屑病、类风湿性关节炎、系统性红斑狼疮和1型糖尿病组成的组中的任一种。
[0066]
11.根据上述8的药物组合物,其中所述组合物用于治疗或预防癌症。
[0067]
12.根据上述11的药物组合物,其中所述癌症选自由黑色素瘤、大肠癌(colon cancer)、肝癌、胶质细胞瘤、卵巢癌、大肠癌、头颈癌、膀胱癌、肾细胞癌、胃癌、乳腺癌、转移癌、前列腺癌、胆囊癌、胰腺癌、血癌、皮肤癌和肺癌组成的组。
[0068]
13.一种用于治疗自身免疫性疾病的方法,其包括对有需要的受试者给予根据上述1至7中任一项的化合物、其立体异构体或药学上可接受的盐。
[0069]
14.根据上述13的用于治疗自身免疫性疾病的方法,其中自身免疫性疾病选自由多发性硬化、炎症性肠病、移植物抗宿主病、哮喘、特应症、银屑病、类风湿性关节炎、系统性红斑狼疮和1型糖尿病组成的组。
[0070]
15.一种用于诱导ahr活性的方法,其包括对有需要的受试者给予根据上述1至7中任一项的化合物、其立体异构体或药学上可接受的盐。
[0071]
16.一种用于抑制il-6的产生的方法,其包括对有需要的受试者给予根据上述1至7中任一项的化合物、其立体异构体或药学上可接受的盐。
[0072]
17.一种用于治疗癌症的方法,其包括对有需要的受试者给予根据上述1至7中任一项的化合物、其立体异构体或药学上可接受的盐。
[0073]
18.根据上述17的用于治疗癌症的方法,其中所述癌症选自由黑色素瘤、大肠癌、肝癌、胶质细胞瘤、卵巢癌、大肠癌、头颈癌、膀胱癌、肾细胞癌、胃癌、乳腺癌、转移癌、前列腺癌、胆囊癌、胰腺癌、血癌、皮肤癌和肺癌组成的组。
[0074]
发明的效果
[0075]
根据本发明的新型化合物、其立体异构体或药学上可接受的盐可以诱导作为免疫调节转录因子的ahr的活性,由此取得不仅控制炎症而且还修复免疫平衡和受损组织的效果。
[0076]
根据本发明的新型化合物、其立体异构体或药学上可接受的盐可以抑制作为炎性因子的il-6的产生,由此取得调节过度免疫应答特别是自身免疫应答的效果。
[0077]
根据本发明的新型化合物、其立体异构体或药学上可接受的盐可以表现出诱导调节性t细胞(treg)的活性的效果。
[0078]
此外,根据本发明的新型化合物、其立体异构体或药学上可接受的盐可以通过调节以上炎性因子而表现出预防和治疗自身免疫性疾病的效果。
附图说明
[0079]
图1和图2示出cyp1a1表达水平的测量以确认本发明的化合物在细胞培养条件下为ahr配体。
[0080]
图3和图4示出本发明的化合物对炎性因子il-6产生的抑制效果的测量。
[0081]
图5示出本发明的化合物的foxp3 调节性t细胞产生效果的测量。
[0082]
图6和图7示出本发明的化合物在患有葡聚糖硫酸钠(dss)诱导的炎症性肠病的动物模型中的炎症性肠病治疗效果。具体地,图6表明,与对照(溶媒)相比,严重性指数越低,治疗完成得越多,而图7显示,大肠的重量与其长度相比越小,炎症性肠病的治疗效果越高。
[0083]
图8和图9示出本发明的化合物在患有dss诱导的炎症性肠病的动物模型中抑制炎性因子(il-1β、il-6、il-17a和tnf-α)的表达和增加免疫调节因子(il-10和foxp3)的表达
的效果。
[0084]
图10示出本发明的化合物在使用fitc-葡聚糖的患有dss诱导的炎症性肠病的动物模型中的黏膜愈合效果。本文中,这意味着检出程度越低,黏膜愈合效果越高。
[0085]
图11示出本发明的化合物在aom/dss结直肠癌动物模型中预防炎症诱发的大肠癌的效果。本文中,这意味着每大肠肿瘤数越少,其预防大肠癌越有效。
[0086]
图12示出本发明的化合物在实验自身免疫性脑脊髓炎(eae)动物模型中治疗多发性硬化的效果。具体地,为了确认多发性硬化的治疗效果,严重性指数作为随时间变化的图示出。本文中,这意味着,与对照相比,严重性指数越低,治疗完成得越多。
[0087]
图13和图14示出本发明的化合物在图12的eae动物模型中抑制炎性因子(ifn-γ、il-17a和il-1β)的表达和增加免疫调节因子(il-10和foxp3)的表达的效果。
[0088]
图15为示出为确认在患有肺移植物抗宿主病的动物模型中移植物抗宿主病(gvhd)的治疗效果而测量的严重性指数的图。
[0089]
图16示出在图15的动物模型中通过本发明的化合物的il-6、il-17a和il-10因子的表达水平的测量。
具体实施方式
[0090]
下文中,将详细描述本发明。
[0091]
除非另有定义,否则在本发明中使用的所有技术术语以与本领域技术人员在本发明的相关领域中通常可以理解的含义相同的含义来使用。此外,虽然在本说明书中将描述优选的方法或样品,但是相似或等同的那些也包括在本发明的范围内。
[0092]
本发明涉及由下式1表示的化合物、其立体异构体或药学上可接受的盐:
[0093]
[式1]
[0094][0095]
在上式中,如果一个位点需要取代基但是在该位点未指明任何取代基,则这意味着省略了氢取代基,这将适用于本发明中的所有结构式。
[0096]
在上式中,r1至r4可以各自独立地为氢或卤素,并且具体地为氢、氟或氯,但是它们不限于此。
[0097]
在上式中,r5和r6可以各自独立地为氢或c
1-c5烷基,具体为氢、甲基或乙基,并且更具体为氢或甲基,但是它们不限于此。
[0098]
在上式中,a可以为c
5-c
12
的单环状基团或双环状基团,并且具体为环戊-1,3-二烯、苯、环己烷、茚、4,5,6,7-四氢茚、萘、1,2,3,4-四氢萘、1,6-二氢并环戊二烯(dihydropentalene)等,但是不限于此。
[0099]
环状基团的各环可以由1至3个杂原子取代,并且,例如,1至3个杂原子可以各自独
立地由n、s、o等取代,但是它们不限于此。杂原子意指碳或氢以外的原子。
[0100]
此外,具体地,杂原子可以取代的位点可以包括以下列出的结构中的q1至q
15
,但是不限于此。
[0101][0102]
在上式中,如果q4为n,则在q4位点无法存在进一步取代,因此,这可以为其中r
11
不存在的情况。
[0103]
环状基团可以由卤素、c
1-c5烷基或c
1-c5烷氧基,例如,f、cl、甲基、乙基、甲氧基、乙氧基等取代,但是不限于此。
[0104]
环状基团的可以由卤素、c
1-c5烷基或c
1-c5烷氧基取代的位点可以具体包括r7至r
30
,但是不限于此。
[0105]
根据本发明的实施方案,a可以选自以下环状基团。
[0106][0107]
(其中r7至r
30
各自独立地为氢、卤素、c
1-c3烷基或c
1-c3烷氧基)。
[0108]
根据本发明的实施方案,a可以具体选自以下环状基团。
[0109][0110]
(其中r7至r
24
各自独立地为氢、卤素、c
1-c3烷基或c
1-c3烷氧基)。
[0111]
此外,根据本发明的实施方案,a可以更具体地选自以下环状基团。
[0112][0113]
(其中r9至r
16
各自独立地为氢、卤素、c
1-c3烷基或c
1-c3烷氧基)。
[0114]
下表1示出由式1表示的化合物的结构的实例,其通过r1至r6与a的组合来具体定
义。
[0115]
[表1]
[0116]
[0117]
[0118][0119]
本发明涉及选自由以下化合物组成的组中的化合物及其立体异构体或药学上可接受的盐。
[0120]
n-(5-溴-6-甲基吡啶-2-基)-2-(1-甲基-1h-吲哚-3-基)乙酰胺;
[0121]
n-(5-溴-6-甲基吡啶-2-基)-2-(1h-吲哚-3-基)乙酰胺;
[0122]
n-(5-溴-6-甲基吡啶-2-基)-2-(5-氯-1h-吲哚-3-基)乙酰胺;
[0123]
n-(苯并[二]噻唑-2-基)-2-(1h-吲哚-3-基)乙酰胺;
[0124]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1h-吲哚-3-基)乙酰胺;
[0125]
n-(5-氯-6-氟吡啶-2-基)-2-(1h-吲哚-3-基)乙酰胺;
[0126]
2-(5-氯-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺;
[0127]
2-(1h-吲哚-3-基)-n-(3,4,5-三甲氧基苯基)乙酰胺;
[0128]
2-(5-氯-1h-吲哚-3-基)-n-(3,4,5-三甲氧基苯基)乙酰胺;
[0129]
n-(3,5-二氯苯基)-2-(1h-吲哚-3-基)乙酰胺;
[0130]
2-(5-氯-1h-吲哚-3-基)-n-(3,5-二氯苯基)乙酰胺;
[0131]
n-(5-溴-6-甲基吡啶-2-基)-2-(5-氟-1h-吲哚-3-基)乙酰胺;
[0132]
2-(5-氯-1h-吲哚-3-基)-n-(吡啶-4-基)乙酰胺;
[0133]
n-(苯并[二]噻唑-2-基)-n-甲基-2-(1-甲基-1h-吲哚-3-基)乙酰胺;
[0134]
n-(苯并[二]噻唑-2-基)-2-(1h-吲哚-3-基)-n-甲基乙酰胺;
[0135]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1h-吲哚-3-基)-n-甲基乙酰胺;
[0136]
n-(5-氯-6-氟吡啶-2-基)-2-(1h-吲哚-3-基)-n-甲基乙酰胺;
[0137]
2-(5-氯-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)-n-甲基乙酰胺;
[0138]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1-甲基-1h-吲哚-3-基)-n-甲基乙酰胺;
[0139]
n-(5-氯-6-氟吡啶-2-基)-n-甲基-2-(1-甲基-1h-吲哚-3-基)乙酰胺;
[0140]
2-(5-氯-1-甲基-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)-n-甲基乙酰胺;
[0141]
2-(5-氯-1-甲基-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺;
[0142]
n-(苯并[二]噻唑-2-基)-2-(1-甲基-1h-吲哚-3-基)乙酰胺;
[0143]
n-(苯并[二]噻唑-2-基)-2-(5-氟-1h-吲哚-3-基)-n-甲基乙酰胺;
[0144]
n-(苯并[二]噻唑-2-基)-2-(5-氯-1-甲基-1h-吲哚-3-基)乙酰胺;
[0145]
n-(苯并[二]噻唑-2-基)-2-(5-氟-1h-吲哚-3-基)乙酰胺;
[0146]
n-(苯并[二]噻唑-2-基)-2-(6-氯-1h-吲哚-3-基)乙酰胺;
[0147]
2-(5-氯-1h-吲哚-3-基)-n-(噻唑-2-基)乙酰胺;
[0148]
2-(5-氯-1h-吲哚-3-基)-n-(喹啉-2-基)乙酰胺;和
[0149]
2-(5-氯-1h-吲哚-3-基)-n-(4,5,6,7-四氢苯并[二]噻唑-2-基)乙酰胺;
[0150]
此外,本发明涉及包含所述化合物、其立体异构体或药学上可接受的盐的药物组合物。
[0151]
药物组合物可以为用于治疗或预防自身免疫性疾病的药物组合物。具体地,所述疾病可以为多发性硬化(ms)、炎症性肠病(ibd)、移植物抗宿主病(gvhd)、哮喘、特应症、银屑病、类风湿性关节炎(ra)、系统性红斑狼疮(sle)、1型糖尿病(t1d)、白塞病或干燥综合征。更具体地,所述疾病可以为多发性硬化、炎症性肠病、移植物抗宿主病、哮喘、特应症、银屑病、类风湿性关节炎、系统性红斑狼疮、1型糖尿病,但是不限于此。
[0152]
在本发明中,“自身免疫性疾病”可以通过体液免疫、细胞免疫或这二者对细胞或组织造成损伤。即,自身免疫性疾病为免疫系统对自身抗原产生不适当的反应从而全身性
地或者在特定的器官中特异性地诱导自身免疫应答等的疾病,其可能会引起慢性炎症。
[0153]“多发性硬化”在广义上是指引起脱髓鞘和瘢痕形成作为体征和症状的炎性疾病,其由大脑和脊髓的轴突周围的脂肪髓鞘的损伤和/或消耗引起。多发性硬化的类型可以包括复发缓解型多发性硬化(recurrent palliative multiple sclerosis,rrms)、继发性进行性多发性硬化(secondary progressive multiple sclerosis,spms)、原发性进行性多发性硬化(primary progressive multiple sclerosis,ppms)和进行性复发性多发性硬化(progressive recurrent multiple sclerosis,prms),但是它们不限于此。
[0154]“炎症性肠病”是指肠道中的异常慢性炎症反复改善和复发的疾病,并且可以对应于选自由克罗恩病、溃疡性大肠炎和肠型白塞病组成的组中的一者,但是不限于此。
[0155]“移植物抗宿主病”为造血干细胞移植期间输注的淋巴细胞攻击具有降低的免疫功能的宿主而引起例如发热、皮疹和肝功能异常等症状的疾病,并且可以侵袭皮肤、肺、肠或肝脏等,但是不限于此。
[0156]“哮喘”是指当暴露于特定的致病物时由于支气管的炎症而反复出现例如咳嗽和呼吸困难等症状的疾病,并且可以由感染、吸烟、过敏原等引起,但是不限于此。
[0157]“特应症”是指特异反应性皮炎(atopic dermatitis),并且为作为慢性复发性炎性皮肤疾病出现例如瘙痒和皮肤干燥等症状的代表性过敏性疾病。
[0158]“银屑病”是指由于免疫系统中的异常而在皮肤或关节中发生的炎性疾病,并且会引起例如出现外观丑陋、增加的角质或红斑斑块并且伴有疼痛等问题。银屑病可以包括选自银屑病性关节炎(psoriatic arthritis)、滴状银屑病(guttate psoriasis)、脓疱性银屑病(pustular psoriasis)、红斑银屑病(red skin psoriasis)、头皮银屑病(scalp psoriasis)、甲银屑病(nail psoriasis)和起止点炎(enthesitis)中的任意一种或多种疾病。
[0159]“类风湿性关节炎”是指特征在于关节部位的慢性炎症的全身性自身免疫性疾病。
[0160]“系统性红斑狼疮”也称为“狼疮”,并且是指作为慢性炎性自身免疫性疾病的、侵袭身体的各种器官例如结缔组织、皮肤、关节、血液和肾脏的全身性疾病。确切原因尚不清楚,但是根据先前的研究,已知遗传因素与该疾病的发生有关。为了帮助诊断狼疮,美国风湿病学会(acr)公布了11种症状、体征和测试结果以帮助将该疾病与其它疾病相区分。根据公开的研究,如果出现11种症状中的4种以上,则可以诊断为狼疮。
[0161]“1型糖尿病”为分泌胰岛素的β细胞被自身免疫反应破坏的免疫介导的疾病。该疾病的原因可以包括特异性靶向分泌胰岛素的β细胞的许多遗传因素和环境因素。该疾病可能会伴有免疫细胞对胰岛的进行性炎性浸润。
[0162]
在本发明中,除了作为本发明的化合物的活性成分以外,还可以使用药学上合适且生理学上可接受的添加剂来制备药物组合物。可以将组合物给予哺乳动物。作为上述添加剂,例如,可以使用赋形剂、崩解剂、甜味剂、粘合剂、包衣剂、膨松剂、润滑剂、助流剂或调味剂。
[0163]
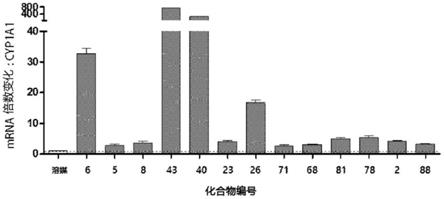
此外,可以优选将本发明的药物组合物配制为除了包含用于给药的上述药学有效量的活性成分以外还包含至少一种药学上可接受的载体的药物组合物。
[0164]“药学有效量”意指足以以适用于药物治疗的合理获益/风险比治疗疾病的量,并且可以基于患者的疾病的类型和严重程度、药物活性、药物敏感性、给药时间、给药途径和
排泄率、治疗持续时间、包括同时使用的药物在内的因素、以及医学领域公知的其它因素来确定有效剂量水平。根据本发明的药物组合物可以作为单独的治疗剂给药或者与其它治疗剂组合给药。此外,组合物可以与常规治疗剂依次或同时给药,并且可以以单剂量或多剂量给药。考虑到所有上述因素,重要的是给予能够取得最大效果而没有副作用的最小量,这样的量可以由本领域技术人员容易地确定。
[0165]
具体地,根据本发明的药物组合物的有效量可以取决于患者的年龄、性别、病况和/或体重、活性成分在体内的吸收、灭活率和排泄率、疾病类型和要组合使用的药物而变化。通常,可以每天或每隔一天给药每1kg体重0.001至150mg、优选0.01至100mg,或者可以分为每天1至3次。然而,剂量可以取决于给药途径、肥胖的严重程度、性别、体重、年龄等而增加或减少,因此,将不会以任何方式限制本发明的范围。
[0166]
此外,“药学上可接受的”是指生理上可接受并且在给予人类时通常不会引起例如胃肠道病症和头晕等过敏反应或类似反应的组合物。
[0167]
载体、赋形剂和稀释剂的实例可以包括乳糖、葡萄糖、蔗糖、山梨糖醇、甘露糖醇、木糖醇、赤藓糖醇、麦芽糖醇、淀粉、阿拉伯树胶、藻酸盐、明胶、磷酸钙、硅酸钙、纤维素、甲基纤维素、聚乙烯吡咯烷酮、水、羟基苯甲酸甲酯、羟基苯甲酸丙酯、滑石、硬脂酸镁和矿物油。此外,可以额外包含填充剂、抗聚集剂、润滑剂、润湿剂、调味剂、乳化剂和防腐剂。
[0168]
此外,可以使用本领域已知的任何方法来配制本发明的组合物,从而在对包括人类的、需要使用本发明的药物组合物的治疗的受试者给药之后提供活性成分的快速、持续或延迟释放。制剂可以为散剂、颗粒剂、片剂、乳剂、糖浆剂、气雾剂、软明胶胶囊剂或硬明胶胶囊剂、无菌注射液、无菌粉末。
[0169]
本发明可以涉及用于治疗自身免疫性疾病的方法,其包括对有需要的受试者给予所述化合物、其立体异构体或药学上可接受的盐。
[0170]
此外,本发明可以涉及用于诱导ahr的活性的方法,其包括给予所述化合物、其立体异构体或药学上可接受的盐。
[0171]
具体地,本发明的化合物可以靶向作为本发明的免疫调节转录因子的芳烃受体(ahr),并且可以用作诱导ahr活性的药剂,由此控制炎症、调节免疫平衡和修复受损组织。因此,化合物可以用于治疗目的,但是不限于此。现有的配体是有毒性的,具有低亲和力和结构稳定性以及高的靶向非特异性(target non-specificity),这带来这些化合物不适合开发成药物组合物的问题。另一方面,当通过具有“药物样性质(drug-like properties)”的本发明的化合物来诱导ahr活性时,其可以有效地用于治疗和预防自身免疫性疾病。
[0172]
本发明可以涉及用于抑制il-6的产生的方法,其包括给予所述化合物、其立体异构体或药学上可接受的盐。
[0173]
具体地,由于已知il-6(一种炎性因子)会引起自身免疫性疾病,因此,本发明的化合物可以通过抑制il-6的产生的机制用于治疗自身免疫性疾病。实际上,存在许多已知的靶向il-6的抑制的用于自身免疫性疾病的治疗剂、以及相关论文。根据以下实验数据,本发明的化合物也被证实抑制il-6的产生并且因此有望具有降低自身免疫应答的效果,由此,本发明的组合物可以用于治疗和预防自身免疫性疾病。
[0174]
此外,本发明涉及用于预防或治疗癌症的组合物,其包含所述化合物、其立体异构体或药学上可接受的盐。
[0175]
在本发明中,“癌症”泛指宿主自身细胞的不受控制的异常生长,其侵袭宿主中最初的异常细胞生长部位的周围组织和远离这些部位的潜在组织。此外,可以包括作为上皮组织(例如,皮肤、鳞状细胞)的癌症的癌(carcinoma);作为结缔组织(例如,骨、软骨、脂肪、肌肉、血管等)的癌症的肉瘤;作为造血组织(例如,骨髓组织)的癌症的白血病;作为免疫细胞的癌症的淋巴瘤和骨髓瘤;中枢神经系统的癌症,包括大脑和脊髓组织中的癌症。
[0176]
具体地,癌症可以选自由黑色素瘤、大肠癌、肝癌、胶质细胞瘤、卵巢癌、大肠癌、头颈癌、膀胱癌、肾细胞癌、胃癌、乳腺癌、转移癌、前列腺癌、胆囊癌、胰腺癌、血癌、皮肤癌和肺癌组成的组,但是不限于此。
[0177]
本发明涉及用于治疗癌症的方法,其包括对有需要的受试者给予所述化合物、其立体异构体或药学上可接受的盐。
[0178]
治疗方法可以包括对被诊断为患有癌症的患者在化疗的任何阶段给予所述化合物、其立体异构体或药学上可接受的盐,并且不限于特定阶段。
[0179]
此外,所述化合物、其立体异构体或药学上可接受的盐可以以前述药物组合物的形式给药,但是不限于此。
[0180]
根据本发明的由式1表示的化合物可以通过各种文献中已知的任何方法来制备。在以下制备例中,简要地描述了表1中列出的一些化合物的合成方法,然而,它们不限于此。
[0181]
下文中,将通过本发明的制备例和实施例详细地描述本发明。
[0182]
制备例
[0183]
1.n-(5-溴-6-甲基吡啶-2-基)-2-(5-氯-1h-吲哚-3-基)乙酰胺(化合物8)的合成
[0184]
[流程1]
[0185][0186]
在室温下搅拌2-(5-氯-1h-吲哚-3-基)乙酸(1.00g,4.77mmol)在ch2cl2(30ml)中的溶液的同时,依次滴加5-溴-6-甲基吡啶-2-胺(892mg,4.77mmol)、1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑[4,5-b]吡啶鎓-3-氧化物六氟磷酸盐(hatu,2.18g,5.72mmol)和三甲胺(1.33ml,9.54mmol)。将反应混合物在室温下搅拌3天,并且将蒸馏水(10ml)添加至混合物中以终止反应。将各层分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱(sio2,己烷:etoac=4:1-2:1)纯化以得到浅灰色化合物(970mg,54%)。
[0187]1h nmr(cdcl3,400mhz):δ8.67(br s,1h),8.00(d,j=8.0hz,1h),7.97(br s,1h),7.55(d,j=4.0hz,1h),7.23(d,j=8.0hz,1h),7.14(m,2h),3.84(s,2h),2.45(s,3h).
[0188]
2.n-(苯并[二]噻唑-2-基)-2-(5-氯-1h-吲哚-3-基)乙酰胺(化合物40)的合成
[0189]
[流程2]
[0190][0191]
除了将制备例1的胺改变为苯并[二]噻唑-2-胺以外,通过与制备例1中相同的实验过程获得白色的标题化合物(1.31g,80%)。
[0192]1h nmr(dmso-d6,400mhz):δ12.58(br s,1h),11.20(br s,1h),7.95(m,1h),7.74(d,j=8.0hz,1h),7.68(d,j=4.0hz,1h),7.43(ddd,j=8.0,8.0,2.0hz,1h),7.39(m,2h),7.29(ddd,j=8.0,8.0,2.0hz,1h),7.09(dd,j=8.0,4.0hz,1h),3.91(s,2h).
[0193]
3. 2-(5-氯-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺(化合物26)的合成
[0194]
[流程3]
[0195][0196]
除了将制备例1的胺改变为5-氯-6-氟吡啶-2-胺以外,通过与制备例1中相同的实验过程获得浅黄色的标题化合物(560mg,35%)。
[0197]1h nmr(cdcl3,400mhz):δ8.38(br s,1h),8.14(dd,j=8.0,2.0hz,1h),7.87(br s,1h),7.77(dd,j=8.0,8.0hz,1h),7.54(dd,j=4.0,2.0hz,1h),7.33(dd,j=8.0,0.8hz,1h),7.21(m,2h),3.87(s,2h).
[0198]
4.n-(5-溴-6-甲基吡啶-2-基)-2-(5-氟-1h-吲哚-3-基)乙酰胺(化合物2)的合成
[0199]
[流程4]
[0200][0201]
除了将制备例1的乙酸改变为2-(5-氯-1h-吲哚-3-基)乙酸以外,通过与制备例1中相同的实验过程获得浅棕色的标题化合物(107mg,44%)。
[0202]1h nmr(cdcl3,400mhz):δ9.09(br s,1h),8.20(br s,1h),8.02(d,j=8.0hz,1h),7.76(d,j=8.0hz,1h),7.20(dd,j=8.0,4.0hz,1h),7.13(dd,j=8.0,4.0hz,1h),6.98(d,j=4.0hz,1h),6.89(m,1h),3.83(s,2h),2.44(s,3h).
[0203]
5.n-(苯并[二]噻唑-2-基)-n-甲基-2-(1-甲基-1h-吲哚-3-基)乙酰胺(化合物45)的合成
[0204]
在室温下、在氩气氛下搅拌n-(苯并[二]噻唑-2-基)-2-(1h-吲哚-3-基)乙酰胺(70.0mg,0.228mmol)在dmf(1ml)中的溶液的同时,滴加t-buok(74.0mg,0.456mmol)并且搅拌5分钟。将mei(28.4μl,0.456mmol)滴加至混合物中并且搅拌30分钟。将蒸馏水(1ml)添加至混合物中以终止反应。将各层分离,并且将有机层用蒸馏水洗涤、经无水mgso4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱(sio2,己烷:etoac=4:1)纯化以得到
氯-1h-吲哚-3-基)乙酰胺来获得白色的标题化合物(26.4mg,54%)。
[0220]1h nmr(cdcl3,500mhz):δ7.83(d,j=8.1hz,1h),7.80(d,j=7.9hz,1h),7.58(d,j=1.0hz,1h),7.43(t,j=7.6hz,1h),7.30(t,j=7.5hz,1h),7.23(d,j=8.7hz,1h),7.20(dd,j=8.7,1.5hz,1h),7.08(s,1h),4.11(s,2h),3.87(s,3h),3.75(s,3h).
[0221]
13
c nmr(cdcl3,125mhz):δ171.51,160.22,148.12,135.51,133.56,129.19,128.65,126.09,125.58,124.03,122.55,121.42,121.23,118.28,110.71,105.57,35.73,33.16,32.49.
[0222]
9.n-(5-氯-6-氟吡啶-2-基)-n-甲基-2-(1-甲基-1h-吲哚-3-基)乙酰胺(化合物36)的合成
[0223]
通过与制备例5中相同的实验过程、同时还使用n-(5-氯-6-氟吡啶-2-基)-2-(1h-吲哚-3-基)乙酰胺来获得黄色的标题化合物(7.2mg,54%)。
[0224]1h nmr(cdcl3,500mhz):δ7.69(t,j=8.7hz,1h),7.50(d,j=7.9hz,1h),7.35(d,j=7.0hz,1h),7.29(d,j=8.2hz,1h),7.23(t,j=7.6hz,1h),7.11(t,j=7.4hz,1h),6.95(s,1h),3.96(s,2h),3.75(s,3h),3.43(s,3h).
[0225]
13
c nmr(cdcl3,125mhz):δ172.27,157.85,155.94,151.78,151.69,141.87,141.86,136.99,127.65,127.61,122.00,119.36,118.82,118.03,117.98,112.96,112.72,109.46,107.06,35.56,32.87,32.82.
[0226]
10. 2-(5-氯-1-甲基-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)-n-甲基乙酰胺(化合物37)的合成
[0227]
通过与制备例5中相同的实验过程、同时还使用2-(5-氯-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺来获得黄色的标题化合物(17.7mg,56%)。
[0228]1h nmr(cdcl3,500mhz):δ7.74(t,j=8.7hz,1h),7.44(s,1h),7.35(br s,1h),7.19(d,j=8.6hz,1h),7.15(dd,j=8.7,1.4hz,1h),6.98(s,1h),3.90(s,2h),3.72(s,3h),3.43(s,3h).
[0229]
13
c nmr(cdcl3,125mhz):δ171.82,157.89,155.98,151.69,151.64,142.02,141.98,135.39,129.12,128.67,125.28,122.27,118.36,117.98,117.94,113.23,112.96,110.54,106.88,35.61,33.06,32.43.
[0230]
11. 2-(5-氯-1-甲基-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺(化合物38)的合成
[0231]
通过与制备例5中相同的实验过程、同时还使用2-(5-氯-1h-吲哚-3-基)-n-(5-氯-6-氟吡啶-2-基)乙酰胺来获得白色的标题化合物(8.20mg,27%)。
[0232]1h nmr(cdcl3,500mhz):δ8.14(d,j=8.5hz,1h),7.86(s,1h),7.76(t,j=8.8hz,1h),7.51(s,1h),7.27(d,j=8.2hz,1h),7.22(dd,j=8.7,1.5hz,1h),7.09(s,1h),3.84(s,2h),3.81(s,3h).
[0233]
13
c nmr(cdcl3,125mhz):δ169.98,157.71,155.80,147.54,147.44,142.93,135.85,129.96,128.47,126.07,123.10,118.22,111.79,111.75,111.00,110.94,110.68,105.82,34.50,33.27
[0234]
12.n-(5-溴-6-甲基吡啶-2-基)-2-(1-甲基-1h-吲哚-3-基)乙酰胺(化合物6)的合成
[0235]
[流程5]
[0236][0237]
在室温下搅拌2-(1-甲基-1h-吲哚-3-基)乙酸(200mg,1.06mmol)在dmf(5ml)中的溶液的同时,依次添加5-溴-6-甲基吡啶-2-胺(197mg,1.06mmol)、1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑[4,5-b]吡啶鎓-3-氧化物六氟磷酸盐(hatu,402mg,1.06mmol)和三甲胺(0.3ml,2.11mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(120mg,31%)。
[0238]1h nmr(cdcl3,400mhz):δ7.99(m,2h),7.72(d,1h,j=12.0hz),7.58(d,1h,j=12.0hz),7.35(d,1h,j=8.0hz),7.27(m,1h),7.15(m,1h),7.06(s,1h),3.87(s,2h),3.80(s,3h),2.42(s,3h)
[0239]
13.n-(5-溴-6-甲基吡啶-2-基)-2-(1h-吲哚-3-基)乙酰胺(化合物5)的合成
[0240]
[流程6]
[0241][0242]
除了将制备例12的乙酸改变为2-(1h-吲哚-3-基)乙酸以外,通过与制备例12中相同的实验过程获得标题化合物(110mg,30%)。
[0243]1h nmr(cdcl3,400mhz):δ8.37(s,1h),7.99(d,1h,j=12.0hz),7.74(d,1h,j=8.0hz),7.60(d,1h,j=8.0hz),7.40(d,1h,j=8.0hz),7.25(m,1h),7.16(m,1h),3.87(s,2h),3.90(s,3h),2.43(s,3h)
[0244]
14.n-(苯并[二]噻唑-2-基)-2-(1h-吲哚-3-基)乙酰胺(化合物43)的合成
[0245]
[流程7]
[0246][0247]
在室温下搅拌2-(1h-吲哚-3-基)乙酸(100mg,0.57mmol)在dmf(3ml)中的溶液的同时,依次添加苯并[二]噻唑-2-胺(197mg,0.57mmol)、1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑[4,5-b]吡啶鎓-3-氧化物六氟磷酸盐(hatu,260mg,0.68mmol)和三甲胺(0.16ml,1.14mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(10mg,6%)。
[0248]1h nmr(cdcl3,400mhz):δ8.99(s,1h),8.32(s,1h)7.80(d,1h,j=8.0hz),7.64(d,1h,j=8.0hz),7.56(d,1h,j=12.0hz),7.40(m,2h),7.29(m,3h),7.16(m,1h),4.03(s,2h)
[0249]
15.n-(5-氯-6-氟吡啶-2-基)-2-(1h-吲哚-3-基)乙酰胺(化合物23)的合成
[0250]
[流程8]
[0251][0252]
除了将制备例14的胺改变为5-氯-6-氟吡啶-2-胺以外,通过与制备例14中相同的实验过程获得标题化合物(6mg,3%)。
[0253]1h nmr(cdcl3,400mhz):δ8.14(dd,1h,j=8.0hz and 2.0hz),7.95(s,1h)7.75(m,1h),7.57(d,1h,j=8.0hz),7.44(m,1h),7.24(m,2h),7.16(m,1h),3.92(s,2h)
[0254]
16. 2-(1h-吲哚-3-基)-n-(3,4,5-三甲氧基苯基)乙酰胺(化合物71)的合成
[0255]
[流程9]
[0256][0257]
除了将制备例14的胺改变为3,4,5-三甲氧基苯胺以外,通过与制备例14中相同的实验过程获得标题化合物(10mg,5%)。
[0258]1h nmr(dmso-d6,400mhz):δ7.59(d,1h,j=8.0hz),7.35(d,1h,j=8.0hz),7.25(m,1h)7.07(m,1h),7.00(s,1h),6.97(m,1h),3.71(s,6h),3.69(s,2h),3.59(s,3h)
[0259]
17. 2-(5-氯-1h-吲哚-3-基)-n-(3,4,5-三甲氧基苯基)乙酰胺(化合物68)的合成
[0260]
[流程10]
[0261][0262]
在室温下搅拌2-(5-氯-1h-吲哚-3-基)乙酸(100mg,0.47mmol)在dmf(3ml)中的溶液的同时,依次添加3,4,5-三甲氧基苯胺(87mg,0.47mmol)、1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑[4,5-b]吡啶鎓-3-氧化物六氟磷酸盐(hatu,217mg,0.57mmol)和三甲胺(0.13ml,0.95mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(10mg,5%)。
[0263]1h nmr(dmso-d6,400mhz):δ11.12(s,1h),10.05(s,1h),7.65(d,1h,j=4.0hz),
7.38(s,1h),7.36(s,1h),7.07(m,1h),6.99(s,2h),3.72(s,6h),3.68(s,2h),3.33(s,3h)
[0264]
18.n-(3,5-二氯苯基)-2-(1h-吲哚-3-基)乙酰胺(化合物81)的合成
[0265]
[流程11]
[0266][0267]
除了将制备例14的胺改变为3,5-二氯苯胺以外,通过与制备例14中相同的实验过程获得标题化合物(8mg,5%)。
[0268]1h nmr(cdcl3,400mhz):δ8.50(s,1h),7.60(s,1h),7.55(d,1h,j=8.0hz),7.41(d,1h,j=8.0hz),7.30(d,1h,j=4.0hz),7.24(m,1h),7.16(m,2h),7.01(m,1h),3.86(s,2h)
[0269]
19. 2-(5-氯-1h-吲哚-3-基)-n-(3,5-二氯苯基)乙酰胺(化合物78)的合成
[0270]
[流程12]
[0271][0272]
除了将制备例17的胺改变为3,5-二氯苯胺以外,通过与制备例17中相同的实验过程获得标题化合物(10mg,6%)。
[0273]1h nmr(cdcl3,400mhz):δ8.33(s,1h),7.56(s,1h),7.36(d,1h,j=8.0hz),7.34(d,1h,j=2.0hz),7.30(bs,1h),7.26(m,1h),7.23(m,2h),7.06(m,1h),3.85(s,2h)
[0274]
20. 2-(5-氯-1h-吲哚-3-基)-n-(吡啶-4-基)乙酰胺(化合物88)的合成
[0275]
[流程13]
[0276][0277]
除了将制备例17的胺改变为吡啶-4-胺以外,通过与制备例17中相同的实验过程获得标题化合物(10mg,7%)。
[0278]1h nmr(dmso-d6,400mhz):δ11.15(s,1h),10.49(s,1h),8.40(m,1h),7.63(d,1h,j=2.0hz),7.57(m,1h)7.37(d,1h,j=8.0hz),7.34(d,1h,j=4.0hz),7.07(dd,1h,j=12.0hz and 2.0hz),3.72(s,6h),3.77(s,2h)
[0279]
21.n-(苯并[二]噻唑-2-基)-2-(1h-吲哚-3-基)-n-甲基乙酰胺(化合物44)的合成
[0280]
[流程14]
[0281][0282]
在室温下搅拌2-(1h-吲哚-3-基)乙酸(960mg,5.48mmol)在dmf(35ml)中的溶液的同时,依次添加n-甲基苯并[二]噻唑-2-胺(600mg,3.65mmol)、n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)脲鎓六氟磷酸盐(hbtu,2.77g,7.31mmol)和n,n-二异丙基乙胺(2.55ml,14.61mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(460mg,39%)。
[0283]1h nmr(dmso-d6,400mhz):δ11.02(s,1h),7.93(m,1h),7.79(m,1h)7.57(m,1h),7.38(m,2h),7.31(m,2h),7.08(m,1h),6.98(m,1h),4.23(s,2h),3.84(s,3h)
[0284]
22.n-(苯并[二]噻唑-2-基)-2-(5-氯-1h-吲哚-3-基)-n-甲基乙酰胺(化合物47)的合成
[0285]
[流程15]
[0286][0287]
在室温下搅拌n-甲基苯并[二]噻唑-2-胺(100mg,0.61mmol)在ch2cl2(12ml)中的溶液的同时,添加三乙胺(0.65ml,3.68mmol),然后进一步滴加2-(5-氯-1h-吲哚-3-基)乙酰氯(280mg,1.23mmol)。将反应混合物在室温下搅拌1天。将蒸馏水添加至混合物中以终止反应。将各层分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(30mg,7%)。
[0288]1h nmr(dmso-d6,400mhz):δ11.22(s,1h),7.95(d,1h,j=8.0hz),7.80(d,1h,j=8.0hz),7.66(d,1h,j=4.0hz),7.42(m,3h)7.31(m,1h),7.09(dd,1h,j=8.0hz and 2.0hz),4.25(s,2h),3.87(s,3h)
[0289]
23.n-(苯并[二]噻唑-2-基)-2-(1-甲基-1h-吲哚-3-基)乙酰胺(化合物54)的合成
[0290]
[流程16]
[0291]
[0292]
在室温下搅拌苯并[二]噻唑-2-胺(214mg,1.43mmol)在ch2cl2(15ml)中的溶液的同时,添加三乙胺(0.66ml,4.75mmol),然后进一步滴加2-(1-甲基-1h-吲哚-3-基)乙酰氯(329mg,1.58mmol)。将反应混合物在室温下搅拌1天。将蒸馏水添加至混合物中以终止反应。将各层分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(120mg,23%)。
[0293]1h nmr(cdcl3,400mhz):δ8.96(s,1h),7.80(d,1h,j=8.0hz),7.63(d,1h,j=8.0hz),7.54(d,1h,j=8.0hz),7.30(m,2h)7.16(m,1h),7.10(s,1h),4.01(s,2h),3.83(s,3h)
[0294]
24.n-(苯并[二]噻唑-2-基)-2-(5-氟-1h-吲哚-3-基)-n-甲基乙酰胺(化合物60)的合成
[0295]
[流程17]
[0296][0297]
在室温下搅拌2-(5-氟-1h-吲哚-3-基)乙酸(352mg,1.83mmol)在dmf(12ml)中的溶液的同时,依次添加n-甲基苯并[二]噻唑-2-胺(200mg,1.22mmol)、n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)脲鎓六氟磷酸盐(hbtu,923mg,2.44mmol)和n,n-二异丙基乙胺(0.9ml,4.87mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(140mg,33%)。
[0298]1h nmr(dmso-d6,400mhz):δ11.12(s,1h),7.95(d,1h,j=8.0hz),7.80(d,1h,j=8.0hz),7.37(m,5h)6.93(m,1h),4.23(s,2h),3.86(s,3h)
[0299]
25.n-(苯并[二]噻唑-2-基)-2-(5-氯-1-甲基-1h-吲哚-3-基)乙酰胺(化合物56)的合成
[0300]
[流程18]
[0301][0302]
在室温下搅拌2-(5-氯-1-甲基-1h-吲哚-3-基)乙酸(300mg,1.34mmol)在dmf(13ml)中的溶液的同时,依次添加苯并[二]噻唑-2-胺(161mg,1.07mmol)、n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)脲鎓六氟磷酸盐(hbtu,1.02g,2.68mmol)和n,n-二异丙基乙胺(0.9ml,5.37mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反
应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(170mg,35%)。
[0303]1h nmr(cdcl3,400mhz):δ9.35(s,1h),7.81(d,1h,j=8.0hz),7.65(d,1h,j=8.0hz),7.48(m,1h),7.39(m,1h),7.25(m,3h),7.04(s,1h),3.94(s,2h),3.76(s,3h)
[0304]
26.n-(苯并[二]噻唑-2-基)-2-(5-氟-1h-吲哚-3-基)乙酰胺(化合物64)的合成
[0305]
[流程19]
[0306][0307]
在室温下搅拌2-(5-氟-1h-吲哚-3-基)乙酸(50mg,0.25mmol)在dmf(3ml)中的溶液的同时,依次添加苯并[二]噻唑-2-胺(31mg,0.20mmol)、n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)脲鎓六氟磷酸盐(hbtu,196mg,0.51mmol)和n,n-二异丙基乙胺(0.2ml,1.04mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(10mg,12%)。
[0308]1h nmr(dmso-d6,400mhz):δ12.65(s,1h),11.10(s,1h),7.95(d,1h,j=8.0hz),7.74(d,1h,j=8.0hz),7.38(m,4h),7.28(m,1h),6.93(m,1h),3.90(s,2h)
[0309]
27.n-(苯并[二]噻唑-2-基)-2-(6-氯-1h-吲哚-3-基)乙酰胺(化合物41)的合成
[0310]
[流程20]
[0311][0312]
除了将制备例26的乙酸改变为2-(6-氯-1h-吲哚-3-基)乙酸以外,通过与制备例26中相同的实验过程获得标题化合物(6mg,3%)。
[0313]1h nmr(meod-d4,400mhz):δ11.65(s,1h),11.10(s,1h),7.85(d,1h,j=8.0hz),7.74(d,1h,j=8.0hz),7.58(d,1h,j=8.0hz),7.43(m,2h),7.31(m,2h),7.05(dd,1h,j=8.0hz and 4.0hz),4.87(s,2h)
[0314]
28. 2-(5-氯-1h-吲哚-3-基)-n-(噻唑-2-基)乙酰胺(化合物99)的合成
[0315]
[流程21]
[0316][0317]
在室温下搅拌2-(5-氯-1h-吲哚-3-基)乙酸(125mg,0.49mmol)在dmf(5ml)中的溶液的同时,依次添加噻唑-2-胺(50mg,0.59mmol)、n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)脲鎓六氟磷酸盐(hbtu,378mg,0.99mmol)和n,n-二异丙基乙胺(0.4ml,2.00mmol)。将反应混合物在室温下搅拌3天。将蒸馏水添加至混合物中以终止反应。将各层用乙酸乙酯分离,并且将有机层用蒸馏水洗涤、经无水na2so4干燥并且过滤。在将滤液减压浓缩之后,将浓缩物通过柱色谱纯化以得到标题化合物(19mg,13%)。
[0318]1h nmr(dmso-d6,400mhz):δ12.31(s,1h),11.18(s,1h),7.66(d,1h,j=2.0hz),7.46(d,1h,j=4.0hz),7.38(s,1h),7.36(m,1h),7.18(d,2h,j=4.0hz),7.07(dd,1h,j=8.0hz and 4.0hz),3.84(s,2h)
[0319]
29. 2-(5-氯-1h-吲哚-3-基)-n-(喹啉-2-基)乙酰胺(化合物109)的合成
[0320]
[流程22]
[0321][0322]
除了将制备例28的胺改变为喹啉-2-胺以外,通过与制备例28中相同的实验过程获得标题化合物(18mg,15%)。
[0323]1h nmr(dmso-d6,400mhz):δ11.16(s,1h),10.98(s,1h),8.30(m,1h),7.89(dd,1h,j=8.0hz and 2.0hz),7.82(d,1h,j=4.0hz),7.72(m,2h),7.48(m,1h),7.40(d,1h,j=2.0hz),7.37(d,1h,j=12.0hz),7.07(dd,1h,j=8.0hz and 4.0hz),3.86(s,2h)
[0324]
30. 2-(5-氯-1h-吲哚-3-基)-n-(4,5,6,7-四氢苯并[二]噻唑-2-基)乙酰胺(化合物104)的合成
[0325]
[流程23]
[0326][0327]
除了将制备例28的胺改变为4,5,6,7-四氢苯并[二]噻唑-2-胺以外,通过与制备例28中相同的实验过程获得标题化合物(19mg,17%)。
[0328]1h nmr(dmso-d6,400mhz):δ12.07(s,1h),11.17(s,1h),7.63(d,1h,j=4.0hz),7.37(d,1h,j=8.0hz and 2.0hz),7.07(dd,1h,j=8.0hz and 4.0hz),3.78(s,2h),2.52(m,8h)
[0329]
以上化合物的结构在表2a至2c中示出,并且化合物的分子量在表2d中列出。
[0330]
[表2a]
[0331][0332]
[表2b]
[0333][0334]
[表2c]
[0335][0336]
[表2d]
[0337]
化合物的编号分子量化合物的编号分子量6358.2388285.735344.2144321.408378.6547355.8443307.3754321.4040341.8160339.3923303.7256355.8426338.1664325.3671340.3741341.8168374.8299291.7681319.19109335.7978353.63104345.852362.20
ꢀꢀ
[0338]
实施例:化合物的活性的测量
–
实验方案
[0339]
1.化合物的搜索和制备
[0340]
为了确认制备的化合物的靶特异性,通过以下方法进行评价。
[0341]
在将在dmem-胎牛血清(fbs)10%培养基中培养的hepg2进行回收然后通过台盼蓝染色确认存活率为97%以上之后,将回收产物在室温下以1200rpm的速度离心5分钟,并且通过将细胞以3
×
105个细胞/ml重悬于dmem-胎牛血清10%培养基中来准备细胞。此后,将细胞以3ml分装至60mm培养皿,并且将各培养皿用在dmem培养基中稀释的50μl浓度为5μm的化合物进行处理,然后在细胞培养箱(5%co2培养箱)中培养24小时。作为对照,使用50μl0.05%二甲亚砜(dmso)/dmem培养基来处理。
[0342]
回收培养的细胞以制备mrna样品。具体地,使用trizol试剂(invitrogen,目录号15596018)通过苯酚-氯仿沉淀法从回收的细胞中提取mrna。从分离的rna,通过逆转录来合成cdna,并且使用cfx96(bio-rad)检测系统中的iq sybr-green supermix(bio-rad)通过实时聚合酶链反应(pcr)来确认cyp1a1的表达。使用gapdh作为对照酶,通过δδct法来比较酶表达水平的相对值。本文中,使用对照来设定一(1)倍。
[0343]
实时聚合酶链反应在58℃的退火温度下、在45次循环的条件下进行,其中使用以下引物序列。
[0344]
人cyp1a1正向,5'-cac cct cat cag taa tgg tca ga-3'(seq id no:1)和反向,5'-aac gtg ctt atc agg acc tc-3'(seq id no:2);人gapdh正向,5'-tga tga cat caa gaa ggt gg-3'(seq id no:3)和反向,5'-tta ctc ctt gga ggc cat gt-3'(seq id no:4)。
[0345]
结果,可以看出受试化合物中,cyp1a1表达水平高于对照(溶媒),并且可以进一步看出显著地诱导了cyp1a1表达(图1和2)。
[0346]
2.对炎性因子il-6的产生的抑制效果
[0347]
为了评估根据本发明的化合物对巨噬细胞il-6的产生的抑制效果,进行以下实验。
[0348]
在将在rpmi-胎牛血清(fbs)10% 2-me(巯基乙醇)培养基中培养的thp-1进行回收并且通过台盼蓝染色确认存活率为97%以上之后,将回收产物在室温下以1200rpm的速度离心5分钟,并且通过将细胞以5
×
105个细胞/ml重悬于rpmi-胎牛血清10% 2-me培养基中来准备细胞。此后,将细胞以500μl分装至24孔板(每个样品3个孔)中,然后,添加0.5μl pma至每孔200ng/ml,并且用在rpmi 2me培养基中稀释的10μl浓度为5μm的化合物处理各孔。在细胞培养箱(5%co2培养箱)中温育48小时之后,以100ng/ml添加5μl溶解于dpbs中的lps用于处理,然后在细胞培养箱(5%co2培养箱)中培养24小时。
[0349]
作为对照,使用10μl 0.05%二甲亚砜(dmso)/rpmi培养基进行处理。使用新的微型管来回收培养的细胞的培养基,并且将细胞用1ml trizol(invitrogen)回收并且储存在-80℃。通过将10μl回收的培养基和40μl分析稀释缓冲液置于facs管(bd falcon)中来将样品稀释至1/5,然后将各样品中的捕获磁珠(capture bead)涡旋,然后,取1μl涡旋的溶液和捕获磁珠稀释液。然后,添加49μl捕获磁珠稀释液以制备50μl每样品的捕获磁珠溶液。在通过涡旋混合捕获磁珠溶液之后,将50μl捕获磁珠溶液置于包含各样品的facs管中,再次涡旋,并且在室温下静置1小时。
[0350]
1小时后,添加1μl pe检测试剂和49μl pe检测试剂稀释液以制备用于各样品的50μl pe检测溶液。涡旋后,将用于各样品的50μl pe检测溶液添加至包含捕获磁珠溶液和样品的facs管中。涡旋后,将facs管在室温下静置1小时。1小时后,将1ml cba洗涤缓冲液添加至各管,以400g离心5分钟,并且除去上清液。轻轻涡旋后,添加150μl固定缓冲液,轻轻涡旋,然后使用流式细胞术进行分析。
[0351]
结果,如图3和图4所示,通过用化合物处理使thp-1通过lps刺激而产生的il-6显著减少。具体地,与对照(溶媒)的结果相比,本发明的所有化合物均显示低的结果,并且这意味着化合物有效地抑制il-6的产生。
[0352]
3.调节性t细胞生成效果
[0353]
为了考察根据本发明的化合物诱导免疫耐受性的效果,如下来进行体外调节性t细胞(foxp3 treg)生成实验。
[0354]
将人外周血(allcells)和磷酸盐缓冲盐水(pbs)以1:1的比例混合以制备混合物,然后将混合物缓慢地放置以使其不混入histopaque(sigma)的上层。在以350g离心20分钟之后,仅收集中间层中的单核细胞层并且用hbss(hanks'平衡盐溶液,)洗涤。在用macs缓冲液(miltenyi biotec)再次洗涤以获得t细胞之后,用cd4微珠(miltenyi biotec)进行阳性选择(automacs分离器,miltenyi biotec)。通过以上方法收集的t细胞通过将细胞以5
×
105个/ml重悬于rpmi-胎牛血清(fbs)10% 2-me(巯基乙醇)培养基中来准备。对于t细胞活化,将10μg/ml抗cd3(ebioscience
tm
)以150μl分装至48孔板中,在细胞培养箱(37℃,5%co2培养箱)中反应3小时,并且用磷酸盐缓冲盐水洗涤以准备板。将250μl重悬的t细胞分装至所准备的板中,并且用2μg/ml的抗cd28(ebioscience
tm
)、5ng/ml的tgfβ-1(r&d systems)和50u/ml的il-2(miltenyi biotec)处理各孔。使用在rpmi 2me培养基中稀释的5μl浓度为2.5μm的各化合物进行处理,然后在细胞培养箱(37℃,5%co2培养箱)中培养7天。作为对照,使用5μl 0.05%二甲亚砜(dmso)/rpmi培养基进行处理。7天后,为了确认生成调节性t细胞的效果,回收培养的细胞以确定foxp3蛋白的存在。
[0355]
将回收的细胞置于5ml facs管(bd falcon)中并且用1ml磷酸盐缓冲盐水洗涤。将细胞重悬于0.1ml的facs缓冲液(0.1%nan3,1%fbs)中并且用1μg人免疫球蛋白g(人igg,sigma)处理以防止抗体的非特异性结合。在4℃下反应15分钟之后,将细胞用facs缓冲液洗涤。然后,将1ml固定/透化溶液(ebioscience
tm
)添加至包含各样品的facs管,然后在4℃下反应1小时。此后,将产物用透化缓冲液(ebioscience
tm
)洗涤两次。然后,使用0.25μg foxp3单克隆抗体(ebioscience
tm
)进行处理,然后将样品染色4至30分钟。将细胞用透化缓冲液洗涤两次,悬浮于0.3ml的facs缓冲液中,并且通过流式细胞术测量。
[0356]
结果,确认到通过用受试化合物no.5、8、43、40、23、26、71、81、2、44、47、56、64和41处理促进foxp3 调节性t细胞的生成(参见图5)。由此可以看出化合物有效地诱导调节性t细胞的生成和增殖。
[0357]
4.炎症性肠病的治疗效果的确认
[0358]
为了研究根据本发明的化合物对炎症性肠病的治疗效果,在c57bl/6小鼠中诱发炎症性肠病,并且如下给予化合物(no.8、43、40、26、44、54和60)以评价功效(图6至7)。
[0359]
在实验的第0天,通过将dss(葡聚糖硫酸钠,mp biomedicals,目录号160110)溶解于1.5%无菌蒸馏水中制备的1.5%dss溶液给予c57bl/6小鼠(8周龄,雌性,18
±
2g)饮用7天。以2天的间隔更换1.5%dss溶液。从实验的第8天起提供无菌蒸馏水用于饮用。从实验的第0天起以2天的间隔测量体重和严重性指数,以确认炎症性肠病的发作。
[0360]
将每只小鼠20mg/kg的化合物完全溶解于对应于给药剂量的10%(v/v)的dmso,然后在cremophor el-磷酸盐缓冲盐水混合物中稀释,以生成最终的dmso:cremophor el:磷酸盐缓冲盐水(1:1:8,v/v/v),然后从实验的第2天至第11天每日口服给药200μl,总计10次。根据分为0至10级的严重性指数体系,以2天的间隔目视观察并且记录炎症性肠病严重性指数。
[0361]
根据以下项目(表3),通过将三个项目的评分求和来评价炎症性肠病症状。
[0362]
[表3]
[0363][0364]
作为分析的结果,确认到溶剂对照组的体重从实验的第6天起开始下降,在实验的第10天下降10%以上,并且100%诱发肠炎,伴有5以上的严重性指数增加。溶剂对照组中的小鼠在严重性指数达到最大的实验的第10天显示严重性指数为7.29
±
2.29。另一方面,在实验的第10天,与溶剂对照组相比,以20mg/kg的本发明的化合物no.8、43、40、26、44、54或60给药的实验组显示在统计学上显著的治疗效果。此外,在实验的第15天的大肠重量:长度比(重量:长度比,mg/cm)的比较证明,在形态学方面,可以显著地抑制肠道炎症(与溶剂对照组相比:*,p《0.05;**,p《0.01;***,p《0.001,参见图6和7)。具体地,本发明的化合物no.8、43、40、26、44、54和60在以20mg/kg的量给药时显示优异的抗炎效果。
[0365]
在实验的第15天,将小鼠的大肠摘出以制备mrna样品。为了提取mrna,将大肠组织用均质器来研磨以获得均质悬浮液。从均质悬浮液,使用easy-spin
tm
(无dna)总rna提取试剂盒(intron biotechnology,目录号17221)通过苯酚-氯仿沉降法提取mrna。从分离的rna,通过逆转录合成cdna,然后使用cfx96(bio-rad)检测系统中的iq sybr-green supermix(bio-rad)通过实时聚合酶链反应(pcr)来确认炎性细胞因子的表达。使用gapdh作为对照酶,通过δδct法来比较酶表达水平的相对值。本文中,使用正常小鼠大肠作为对照来设定一(1)倍。
[0366]
实时聚合酶链反应在58℃的退火温度下、在45次循环的条件下进行,并且使用以下引物序列。
[0367]
小鼠il-1β正向,5'-ctc gtg ctg tcg gac cca tat-3'(seq id no:5)和反向,5'-ttg aag aca aac cgc ttt tcc a-3'(seq id no:6);
[0368]
小鼠il-6正向,5
′‑
cat gtt ctc tgc gaa atc gtg g-3
′
(seq id no:7)和反向,5
′‑
aac gca cta ggt ttg ccg agt a-3
′
(seq id no:8);
[0369]
小鼠il-17a正向,5'-ttt aac tcc ctt ggc gca aaa-3'(seq id no:9)和反向,5'-ctt tcc ctc cgc att gac ac-3'(seq id no:10);
[0370]
小鼠tnf-α正向,5'-cca cac cgt cag ccg att tg-3'(seq id no:11)和反向,5'-cac cca ttc cct tca cag agc-3'(seq id no:12);
[0371]
小鼠il-10正向,5'-caa ggc agt gga gca ggt gaa-3'(seq id no:13)和反向,5'-cgg aga gag gta caa acg agg tt-3'(seq id no:14);
[0372]
小鼠foxp3正向,5'-ccc atc ccc agg agt ctt g-3'(seq id no:15)和反向,5'-acc atg act agg ggc act gta-3'(seq id no:16);
[0373]
小鼠gapdh正向,5'-ttc acc acc atg gag aag gc-3'(seq id no:17)和反向,5'-ggc atg gac tgt ggt cat ga-3'(seq id no:18)。
[0374]
通过给予化合物no.8、40、26或54,与溶剂对照组相比,使大肠病变中炎性细胞因
子il-1β、il-6、il-17a和tnf-α的表达水平显著降低(与溶剂对照组相比:**,p《0.01;***,p《0.001,参见图8)。此外,通过给予化合物no.8、40、26或54,与溶剂对照组相比,使大肠病变中免疫调节因子il-10和foxp3的表达水平显著增加(与溶剂对照组相比:***,p《0.001,参见图9)。由这些结果,可以看出本发明的化合物no.8、40、26和54可以使肠内炎性因子的表达显著降低,同时还使肠内免疫调节因子的表达显著增加。
[0375]
为了研究根据本发明的化合物的黏膜愈合效果,在c57bl/6小鼠中诱发炎症性肠病,并且通过如下给予化合物(no.40、26、54)来评估肠上皮屏障完整性的恢复程度。
[0376]
在实验的第0天,将通过将1.5%dss溶解于无菌蒸馏水中制备的1.5%dss溶液给予c57bl/6小鼠(8周龄,雌性,18
±
2g)饮用7天。以2天的间隔更换1.5%dss溶液。从实验的第8天起提供无菌蒸馏水用于饮用。从实验的第0天起,以2天的间隔测量体重和严重性指数,从而确认炎症性肠病的发作。
[0377]
在根据本发明的化合物no.40、26或54给药组中,将每只小鼠20mg/kg的化合物完全溶解于对应于给药剂量的10%(v/v)的dmso中,然后,在cremophor el-磷酸盐缓冲盐水混合物中稀释以制备最终的dmso:cremophor el:磷酸盐缓冲盐水(1:1:8,v/v/v),然后,从实验的第2天至第9天,每日口服给予200μl所制备的溶液,总计8次。
[0378]
fitc-葡聚糖给药前一天,使小鼠禁水过夜。在实验的第10天,将600mg/kg的fitc-葡聚糖(异硫氰酸荧光素-葡聚糖,sigma aldrich,目录号fd40)在磷酸盐缓冲盐水中稀释并且以200μl口服给予小鼠一次。口服给药后4小时,在从心脏提取的血清中测量荧光(荧光计,激发485-490nm、发射528-530nm)。
[0379]
通过给予化合物no.40、26或54,与溶剂对照组相比,使血清fitc-葡聚糖显著降低(与溶剂对照组相比:***,p《0.001,参见图10)。由该结果,可以看出本发明的化合物no.40、26和54表现出显著的黏膜愈合效果。
[0380]
因此,本发明的化合物8、43、40、26、44、54和60在患有炎症性肠病的小鼠模型中口服给药时具有治疗效果。因此,可以提示这些化合物作为用于炎症性肠病的新型口服给药治疗剂的有用的治疗策略。
[0381]
5.预防炎症诱发的大肠癌的效果的确认
[0382]
为了研究根据本发明的化合物预防炎症诱发的大肠癌的效果,通过如下对在c57bl/6小鼠中的aom/dss诱导的大肠癌小鼠模型分别给予化合物no.40和26来评价医疗功效(图11)。作为致癌物质的氧化偶氮甲烷(aom)在单独给予小鼠时不会引起大肠癌。然而,当使用dss追加炎症时,发生大肠癌。
[0383]
将aom(sigma aldrich,目录号a5486)用生理盐水稀释至浓度为10mg/kg,然后以7天的间隔腹腔内给药3次(实验第0天、第7天、第14天)。在实验的第7天,将通过将1.5%dss溶解于无菌蒸馏水中制备的1.5%dss溶液给予c57bl/6小鼠(8周龄,雌性,18
±
2g)饮用7天。此外,以2天的间隔更换1.5%dss溶液。从实验的第8天起提供无菌蒸馏水用于饮用。
[0384]
在根据本发明的化合物no.40和26的给药组中,分别地,将每只小鼠20mg/kg的化合物完全溶解于对应于给药剂量的10%(v/v)的dmso中,然后在cremophor el-磷酸盐缓冲盐水混合物中稀释以制备最终的dmso:cremophor el:磷酸盐缓冲盐水(1:1:8,v/v/v),然后从实验的第7天至第21天,每日口服给予200μl所制备的溶液,总计14次。在溶剂对照组的体重与达到峰值体重的实验的第56天相比降低15%时,确认到化合物的大肠癌预防效果。
[0385]
在实验的第93天,将小鼠的大肠摘出以确认在大肠中发生肿瘤。大肠中的肿瘤数在溶剂对照组中为11.33
±
4.33,在化合物no.40的情况下为3.00
±
2.00,并且在化合物no.26的情况下为3.17
±
1.17,从而,通过给予化合物no.40和26,与溶剂对照组相比,使肿瘤数显著减少(与溶剂对照组相比:**,p《0.01,参见图11)。
[0386]
因此,本发明的化合物no.40和26对炎症诱发的大肠癌的发生具有抑制效果。因此,可以提示这些化合物作为具有大肠癌预防效果的用于炎症性肠病的新型口服给药的治疗剂的有用的治疗策略。
[0387]
6.多发性硬化治疗效果的确认
[0388]
为了研究根据本发明的化合物对多发性硬化的治疗效果,在c57bl/6小鼠中诱发自身免疫性脑脊髓炎(eae)并且通过给予化合物(8、43、40、26)来评价医疗功效(图12至14)。
[0389]
在第0天,将髓鞘少突胶质细胞糖蛋白35-55(mog 35-55,peptron)(200μg)、热灭活的结核分枝杆菌(difco,目录号231141)(500μg)和佐剂(完全弗氏佐剂,sigma aldrich,目录号f5506)混合在一起,然后浸泡7分钟。在将100μl浸水肽(submerged peptide)皮下注射至各c57bl/6小鼠(7周龄,雌性,17
±
2g)的双侧腹部之后,对尾部静脉内注射给予100μl百日咳毒素(sigma aldrich,目录号p2980)(200ng)。
[0390]
在实验的第2天,静脉内注射给予相同量的百日咳毒素。检查小鼠是否有来自注射部位的浸液渗漏(immersion leaking),并且从实验的第7天起目视观察以确认多发性硬化的发作。
[0391]
在用化合物no.8、43、40和26处理的组中,将每只小鼠20mg/kg的化合物完全溶解于对应于给药剂量的10%(v/v)的dmso中,然后,在cremophor el-磷酸盐缓冲盐水混合物中稀释以制备最终的dmso:cremophor el:磷酸盐缓冲盐水(1:1:8,v/v/v),然后从实验的第12天至第17天每日腹腔内给予200μl所制备的溶液,总计6次。在分为0-5级的严重性指数体系中,从实验的第7天起以2天的间隔目视观察并且记录多发性硬化指数,并且根据以下项目对自身免疫性脑脊髓炎症状进行指数评价(参见表4)。
[0392]
[表4]
[0393]
评分症状0无症状1尾部无力2尾部无力且后肢虚弱3后肢瘫痪4后肢瘫痪且前肢虚弱5死亡或濒临死亡
[0394]
作为分析的结果,确认到所有实验组从实验的第7天起发生多发性硬化,并且在实验的第18天诱发100%的严重性指数为3.0以上的急性反应。
[0395]
溶剂对照组中的小鼠的严重性指数在实验的第18天(为急性反应期)为3.33
±
0.17,并且在实验的第36天(为慢性反应期)为3.33
±
0.17。此外,溶剂对照组在整个实验期间显示复发缓解模式(relapse-remitting pattern)和高的严重性指数。
[0396]
另一方面,化合物治疗组的严重性指数为:在实验的第18天,在化合物no.8治疗组
中为1.17
±
0.56,在化合物no.43治疗组中为1.83
±
0.17,在化合物no.40治疗组中为1.17
±
0.22,并且在化合物no.26治疗组中为1.33
±
0.56;并且,在实验的第36天,在化合物no.8治疗组中为1.17
±
0.56,在化合物no.43治疗组中为2.00
±
0.33,在化合物no.40治疗组中为1.33
±
0.62,在化合物no.26治疗组中为1.33
±
0.22,与溶剂对照组相比,显示缓解急性反应和慢性反应的治疗效果。
[0397]
此外,可以确认用化合物no.8、43、40和26处理的组从实验的第16天起与溶剂对照组相比具有在统计学上显著的治疗效果,并且即使在停止给药后,与溶剂对照组相比也维持在统计学上显著的治疗效果(与溶剂对照组相比:***,p《0.001,参见图12)。从以上结果,可以看出对小鼠给药20mg/kg表现出优异的初始治疗效果和持续的复发预防效果。
[0398]
在实验的第42天,将小鼠的脊髓摘出以制备mrna样品。为了提取mrna,将脊髓组织用均质器研磨以获得均质悬浮液。使用trizol试剂(invitrogen,目录号15596018)通过苯酚-氯仿沉淀法从均质悬浮液提取mrna。然后,通过逆转录从分离的rna合成cdna,并且使用cfx96(bio-rad)检测系统中的iq sybr-green supermix(bio-rad)通过实时聚合酶链反应(pcr)来研究炎性细胞因子的表达。使用gapdh作为对照酶,通过δδct法来比较酶表达水平的相对值。本文中,使用wt小鼠脊髓作为对照来设定一(1)倍。
[0399]
实时聚合酶链反应在58℃的退火温度下、在45次循环的条件下进行,并且使用以下引物序列。
[0400]
小鼠ifn-γ正向,5'-atg aac gct aca cac tgc atc-3”(seq id no:19)和反向,5'-cca tcc ttt tgc cag ttc ctc-3”(seq id no:20);
[0401]
小鼠il-17a正向,5'-ttt aac tcc ctt ggc gca aaa-3”(seq id no:21)和反向,5'-ctt tcc ctc cgc att gac ac-3”(seq id no:22);
[0402]
小鼠il-1β正向,5'-ctc gtg ctg tcg gac cca tat-3”(seq id no:23)和反向,5'-ttg aag aca aac cgc ttt tcc a-3”(seq id no:24);
[0403]
小鼠gapdh正向,5'-ttc acc acc atg gag aag gc-3”(seq id no:25)和反向,5'-ggc atg gac tgt ggt cat ga-3”(seq id no:26);
[0404]
小鼠il-10正向,5'-caa ggc agt gga gca ggt gaa-3'(seq id no:27)和反向,5'-cgg aga gag gta caa acg agg tt-3”(seq id no:28);
[0405]
小鼠foxp3正向,5'-ccc atc ccc agg agt ctt g-3”(seq id no:29)和反向,5'-acc atg act agg ggc act gta-3”(seq id no:30)。
[0406]
此外,通过给予化合物no.8、43、40和26,与溶剂对照组相比,使脊髓病变中炎性细胞因子ifn-γ、il-17a和il-1β各自的表达水平显著降低(与溶剂对照组相比:*,p《0.05;**,p《0.01;***,p《0.001,参见图13)。通过给予化合物no.8、43、40和26,与溶剂对照组相比,使脊髓病变中免疫调节因子il-10和foxp3的表达水平显著增加(与溶剂对照组相比:*,p《0.05;**,p《0.01,参见图14)。
[0407]
因此,本发明的化合物no.8、43、40和26在多发性硬化小鼠模型中具有治疗功效,并且即使在停止给药后,也持续维持预防复发的效果。因此,可以提示这些化合物作为用于多发性硬化的新型口服给药的治疗剂的有用的治疗策略。
[0408]
7.移植物抗宿主病治疗效果的确认
[0409]
为了研究本发明的化合物对移植物抗宿主病(gvhd)的抑制效果,如下在c57bl/6
小鼠中通过同种异体骨髓移植诱发急性移植物抗宿主病,并且给予化合物(no.40或26)以评价其功效(图15和16)。
[0410]
将balb/c ifn-γ敲除小鼠(8至12周龄,雌性,18
±
3g)的脾脏摘出、通过添加rpmi培养基来粉碎然后通过40μm细胞过滤器(bd falcon),由此获得单细胞悬液。将单细胞悬液离心(1200rpm,5分钟),并且在将上清液弃去之后,添加1ml ack(氯化铵/碳酸氢钾)裂解缓冲液(0.15m nh4cl,1mm khco3,0.1mm na2edta),然后搅拌1分钟,然后用rpmi培养基洗涤。
[0411]
离心后,使细胞悬液在小鼠cd90.2微珠(miltenyi biotec,目录号130-121-278)上、在4℃下反应20分钟。在反应完成后,将细胞悬液离心、用10ml运行缓冲液(miltenyi biotec,目录号130-091-221)洗涤、然后用3ml运行缓冲液重悬。然后,使用auto macs pro(miltenyi biotec)从细胞悬液中获得cd90.2
t细胞(阳性选择)。为了获得要与获得的cd90.2
t细胞一起移植的骨髓细胞,无菌地获取野生型balb/c小鼠(8-12周龄,雌性,18
±
3g)的股骨和胫骨二者。将股骨和胫骨的端部切断,并且通过用注射器(股骨21g,胫骨26g)将rpmi培养基灌注至骨组织来提取骨髓。使提取的骨髓通过40μm细胞过滤器以获得单细胞悬液。
[0412]
将骨髓单细胞悬液离心,并且在将上清液弃去之后,添加500μl ack裂解缓冲液,然后搅拌30秒并且用rpmi培养基洗涤溶液。离心后,使悬液在小鼠cd90.2微珠上、在4℃下反应20分钟。在反应完成后,将细胞悬液离心、用10ml运行缓冲液洗涤然后用3ml运行缓冲液重悬。然后,通过auto macs pro从细胞悬液中获得cd90.2-t细胞耗竭的骨髓细胞(tcd-bm)(阴性选择)。用磷酸盐缓冲盐水洗涤获得的ifn-γ敲除cd90.2
t细胞和正常的tcd-bm。通过将t细胞以1
×
107个/ml重悬于磷酸盐缓冲盐水中来制备t细胞,同时通过将tcd-bm以5
×
107个/ml重悬于磷酸盐缓冲盐水中来制备tcd-bm。
[0413]
通过使用辐射辐照器,以3小时的间隔用850cgy的辐射照射正常的c57bl/6小鼠(9至11周龄,雌性,19
±
3g)。将通过将制备的cd90.2
t细胞和tcd-bm以1:1的比例混合来制备的移植物以100μl的量通过c57bl/6小鼠的尾静脉注射。在根据本发明的化合物no.40或26给药组中,将每只小鼠20mg/kg的化合物完全溶解于对应于给药剂量的10%(v/v)的dmso中,然后在cremophor el-磷酸盐缓冲盐水混合物中稀释以制备最终的dmso:cremophor el:磷酸盐缓冲盐水(1:1:8,v/v/v),然后从移植后第4天至第9天每日腹腔内给予溶液200μl,总计6次。在如下严重性指数体系中,通过目视观察以2天的间隔评价移植物抗宿主病严重性指数,所述严重性指数体系分为总计10分,其中每个项目0至2分,包括体重的降低、毛发状况、姿势、活动和皮肤变化。
[0414]
作为分析的结果,在移植后第7天,在溶剂对照组的小鼠中100%诱发移植物抗宿主病,严重性指数为4.12
±
1.20。此外,在移植后第14天,严重性指数为:在溶剂对照组中为6.20
±
1.20;在化合物no.40治疗组中为0.60
±
0.60;并且在化合物no.26治疗组中为2.80
±
0.80,与溶剂对照组相比,显示显著缓解的移植物抗宿主病和治疗效果(与溶剂对照组相比:***,p《0.001,参见图15)。
[0415]
在实验的第15天,将小鼠的肺摘出以制备mrna样品。为了提取mrna,将脊髓组织用均质器研磨以获得均质悬浮液。使用trizol试剂(invitrogen,目录号15596018)通过苯酚-氯仿沉淀法从均质悬浮液中提取mrna。然后,通过逆转录从分离的rna合成cdna,并且使用cfx96(bio-rad)检测系统中的iq sybr-green supermix(bio-rad)通过实时聚合酶链反应
(pcr)来确认炎性细胞因子的表达。使用gapdh作为对照酶,通过δδct法来比较酶表达水平的相对值。本文中,使用正常小鼠脊髓作为对照来设定一(1)倍。
[0416]
实时聚合酶链反应在58℃的退火温度下、在45次循环的条件下进行,并且使用以下引物序列。
[0417]
小鼠il-17a正向,5'-ttt aac tcc ctt ggc gca aaa-3'(seq id no:31)和反向,5'-ctt tcc ctc cgc att gac ac-3'(seq id no:32);
[0418]
小鼠il-6正向,5'-cat gtt ctc tgc gaa atc gtg g-3'(seq id no:33)和反向,5'-aac gca cta ggt ttg ccg agt a-3'(seq id no:34);
[0419]
小鼠il-10正向,5'-caa ggc agt gga gca ggt gaa-3'(seq id no:35)和反向,5'-cgg aga gag gta caa acg agg tt-3'(seq id no:36);
[0420]
小鼠gapdh正向,5'-ttc acc acc atg gag aag gc-3'(seq id no:37)和反向,5'-ggc atg gac tgt ggt cat ga-3'(seq id no:38)。
[0421]
通过给予化合物no.40和26,与溶剂对照组相比,使肺组织中炎性细胞因子il-6和il-17a的表达水平显著降低。此外,通过给予化合物no.40和26,与溶剂对照组相比,使肺组织中免疫调节因子il-10的表达水平显著增加(与溶剂对照组相比:**,p《0.01;***,p《0.001,参见图16)。
[0422]
因此,本发明的化合物no.40和26在患有移植物抗宿主病的小鼠模型中具有治疗功效,并且即使在停止给药后,也由于免疫调节因子的增加而持续维持治疗功效。因此,可以提示这些化合物作为用于移植物抗宿主病的新型口服给药的治疗剂的有用的治疗策略。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。