1.本发明涉及粉末、粉末的制造方法及溶液的制造方法。更具体而言,涉及后述公开的通式(a)所示的氟化双酚类化合物的粉末、该粉末的制造方法、及使用该粉末的溶液的制造方法。
背景技术:
2.以2,2-双(4-羟基苯基)六氟丙烷(以下也简写为“bis-af”)为代表的氟化双酚类化合物经常用作各种树脂的原料等。
3.作为一个例子,在与环氧树脂组合物相关的文献的专利文献1中,记载了:作为制造例1,在乙二醇及纯水的混合溶剂中进行使bis-af结晶化等操作,而得到该白色粉末。
4.作为另一例,专利文献2的实施例等中记载了:利用盐酸进行中和而使溶于碱性水溶液的bis-af析出,得到作为杂质的六氟丙酮减少的bis-af的固体物。
5.作为另一个例子,在专利文献3的权利要求书、实施例等中,记载了一种2,2-双(4-羟基苯基)六氟丙烷的精制法,该精制法通过将2,2-双(4-羟基苯基)六氟丙烷及水加热至常温以上,将溶解液(水溶液)冷却而得到析出的固体。
6.现有技术文献
7.专利文献
8.专利文献1:日本特开2007-246819号公报
9.专利文献2:日本特开平4-054144号公报
10.专利文献3:日本特开平2-067239号公报
技术实现要素:
11.发明要解决的问题
12.以bis-af为代表的氟化双酚类化合物在工业上通常以粉末的形态制造、销售。另外,有时以粉末的形态直接用于各种工业工艺中。
13.根据本发明人等的见解,以往的粉末状的氟化双酚类化合物在工业上的操作性的观点方面存在改善的余地。“工业上的操作性”是指例如以下项目中的1项或2项以上。
14.·
过滤性良好
15.·
对湿润的粉末进行干燥时的干燥时间较短
16.·
不易吸湿
17.·
粉末的流动性良好
18.·
溶剂溶解性良好,具体而言,迅速溶于溶剂中
19.·
不易块状化(结块)
20.本发明是鉴于这样的情况而完成。本发明的目的之一在于提供工业上的操作性优异的粉末状氟化双酚类化合物。
21.用于解决问题的方案
22.本发明人等完成了以下第1发明~第4发明。
23.第1发明如下。
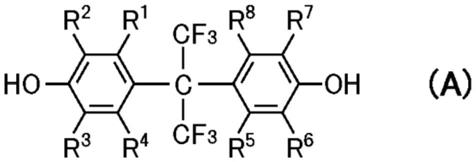
24.一种粉末,其是以下通式(a)所示的化合物的粉末,
25.通过激光衍射散射法测得的众数粒径dm为75~150μm,
26.ca离子的含量低于1ppm。
[0027][0028]
通式(a)中,r1~r8分别独立地表示氢原子、碳数为1~4的烷基、卤素原子或氨基。
[0029]
第2发明如下。
[0030]
一种粉末,其是以下通式(a)所示的化合物的粉末,
[0031]
在将通过激光衍射散射法测得的体积基准累积50%直径设为d
50
、将通过激光衍射散射法测得的算术体积平均直径设为d
ave
时,
[0032]d50
为50~100μm,
[0033]d50
/d
ave
为1.1~1.5。
[0034][0035]
通式(a)中,r1~r8分别独立地表示氢原子、碳数为1~4的烷基、卤素原子或氨基。
[0036]
第3发明如下。
[0037]
一种粉末,其是以下通式(a)所示的化合物的粉末,
[0038]
通过激光衍射散射法测得的体积基准累积50%直径d
50
为50~100μm,
[0039]
所述粉末的休止角为35~49
°
。
[0040][0041]
通式(a)中,r1~r8分别独立地表示氢原子、碳数为1~4的烷基、卤素原子或氨基。
[0042]
第4发明如下。
[0043]
一种粉末,其是2,2-双(4-羟基苯基)六氟丙烷的粉末,
[0044]
x射线衍射光谱中的2θ=22.3
°
附近的峰的半值宽度为0.050
°
以上且0.180
°
以下,
[0045]
x射线衍射光谱中的2θ=23.7
°
附近的峰的半值宽度为0.050
°
以上且0.120
°
以下,
[0046]
x射线衍射光谱中的2θ=25.8
°
附近的峰的半值宽度为0.040
°
以上且0.120
°
以下。
[0047]
另外,本发明人等完成了以下的粉末制法发明。
[0048]
一种粉末的制造方法,其是以下通式(a)所示的化合物的粉末的制造方法,
[0049]
该方法包括如下工序:
[0050]
熔解工序,通过将包含该通式(a)所示的化合物的原料物质、及水系分散介质放入容器中并进行加热,从而使前述原料物质在前述水系分散介质的存在下熔解而得到包含前述原料物质的熔解物及前述水系分散介质的不均匀液体;
[0051]
结晶化工序,通过使前述不均匀液体降温,从而使前述熔解物结晶化,从而得到结晶;
[0052]
在前述熔解工序中,前述原料物质的熔解温度t1下的前述通式(a)所示的化合物在前述水系分散介质中的溶解度为10[g/100g]以下。
[0053][0054]
通式(a)中,r1~r8分别独立地表示氢原子、碳数为1~4的烷基、卤素原子或氨基。
[0055]
另外,本发明人等完成了以下的溶液制法发明。
[0056]
一种溶液的制造方法,其是包含以下通式(a)所示的化合物的溶液的制造方法,
[0057]
该方法包括使用溶剂及第1发明~第4发明的至少任一发明的粉末而得到该通式(a)所示的化合物的溶液的工序。
[0058][0059]
通式(a)中,r1~r8分别独立地表示氢原子、碳数为1~4的烷基、卤素原子或氨基。
[0060]
发明的效果
[0061]
本发明的粉末在工业上的操作性优异。
附图说明
[0062]
图1是用于对第2实施方式的粉末的粒径分布进行说明的图。
[0063]
图2是用于对第2实施方式的粉末的粒径分布进行说明的图。
具体实施方式
[0064]
以下对本发明的实施方式详细进行说明。
[0065]
在本说明书中,数值范围的说明中的“x~y”这一表述,除非特别说明,否则表示x以上且y以下。例如,“1~5质量%”是指“1质量%以上且5质量%以下”。
[0066]
在本说明书的基团(原子团)的表述中,经取代或未经取代的表述包括不具有取代基的情况及具有取代基的情况这两者。例如,“烷基”不仅包括不具有取代基的烷基(未经取代的烷基),还包括具有取代基的烷基(经取代的烷基)。
[0067]
本说明书中的“电子设备”术语以包括半导体芯片、半导体元件、印刷布线基板、电路显示器装置、信息通信终端、发光二极管、物理电池、化学电池等、应用电子工学技术的元
件、装置、最终制品等的含义使用。
[0068]
在本说明书中,有时将通式(a)所示的化合物记为“化合物(a)”。
[0069]
在本说明书中,有时将第1发明的实施方式记为第1实施方式,将第2发明的实施方式记为第2实施方式,将第3发明的实施方式记为第3实施方式,将第4发明的实施方式记为第4实施方式。
[0070]
另外,在本说明书中,有时将前述公开的粉末制法发明的实施方式记为第1制法。另一方面,有时将可制造本发明的粉末(第1发明~第4发明中的至少任一发明的粉末)的粉末的制造方法记为第2制法,但该第2制法不属在前述公开的粉末制法发明。
[0071]
《粉末》
[0072]
第1~第3实施方式的粉末是以下通式(a)所示的化合物(化合物(a))的粉末。另外,第4实施方式的粉末是在以下通式(a)中r1~r8全部为氢原子的化合物的粉末。
[0073][0074]
通式(a)中,r1~r8分别独立地表示氢原子、碳数为1~4的烷基、卤素原子或氨基。
[0075]
通式(a)中,作为r1~r8的碳数为1~4的烷基,可列举出甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基等。
[0076]
通式(a)中,优选为r1~r8分别独立地为氢原子或氨基。
[0077]
通式(a)中,在r1~r8中的至少任一者为除氢原子以外的基团的情况下,该除氢原子以外的基团优选存在于r2、r3、r6、r7中的任意位置。原因在于容易合成化合物。
[0078]
通式(a)中,r1~r8中,优选0~4个为除氢原子以外的基团,更优选0~2个为除氢原子以外的基团。
[0079]
作为化合物(a),具体而言,可列举出2,2双(4-羟基苯基)六氟丙烷、2,2-双(4-羟基-3-甲苯基)六氟丙烷、2,2-双(3-乙基-4-羟基苯基)六氟丙烷、2,2-双(4-羟基-3,5-二甲苯基)六氟丙烷、2,2-双(3-氟-4-羟基苯基)六氟丙烷、2,2-双(3-溴-4-羟基苯基)六氟丙烷、2,2-双(3,5-二溴-4-羟基苯基)六氟丙烷、2,2-双(3-氨基-4-羟基苯基)六氟丙烷等。
[0080]
特别是,可列举出以下的化合物作为优选的化合物(a)。
[0081][0082]
(第1实施方式)
[0083]
第1实施方式的粉末的通过激光衍射散射法测得的众数粒径(mode particle diameter)dm为75~150μm,优选为75~135μm,进一步优选为75~120m。
[0084]
第1实施方式的粉末的ca离子的含量低于1ppm,优选为0.7ppm以下,进一步优选为0.5ppm以下。为了慎重起见事先说明,在本说明书中,ca离子(及下述na离子)的含量的“ppm”是“粉末中的ca离子的质量/粉末的质量”的百万分率。“ppm”与质量%的关系为1ppm=0.0001质量%。
[0085]
第1实施方式的粉末在工业上的操作性良好。具体而言,第1实施方式的粉末具有“过滤性良好”、“可缩短对湿润的粉末进行干燥时的干燥时间”等优点。这些优点推测出原因在于dm为75~150μm。
[0086]
虽详情并不清楚,但推测出或许是由于dm为75μm以上,因此颗粒之间存在适度的“间隙”,液体变得容易流动。另外,推测出由于dm为150μm以下,因此该“间隙”不过大,颗粒之间本来就不易保持大量液体。
[0087]
另外,本发明人等在第1实施方式中将粉末中的ca离子的含量控制在低于1ppm。
[0088]
ca离子是一般工业用水中不可避免地含有的成分。由此,若将ca离子的含量作为指标,以其量低于1ppm的方式制造化合物(a)的粉末,则得到的粉末中的除ca离子以外的各种微量杂质(来源于工业用水)的量也可一并减少。即,认为ca离子的量低于1ppm的化合物(a)中,不仅ca离子较少,各种杂质也较少,该化合物(a)可适宜用于各种技术领域。此外,如下所述,ca离子可通过离子色谱法进行测定,因此,也可使量产化设备中的工序管理变得容易。
[0089]
顺便一提,ca离子的含量低于1ppm直接意味着第1实施方式的粉末可优选用作要求金属离子量较少的电子设备制造用的材料。
[0090]
ca离子的含量优选为0.7ppm以下,更优选为0.5ppm以下。
[0091]
ca离子基本上越少越优选。ca离子的含量也可以为零(装置的测定极限以下)。从实用性的观点出发,ca离子的含量例如为0.01ppm以上。
[0092]
用于制造第1实施方式的粉末的方法及条件没有限定。通过选择适当的方法及条件,可以得到dm为75~150μm、ca离子的含量低于1ppm的化合物(a)的粉末。优选地,如下述的第1制法所述,优选使原料物质在水系分散介质中熔融(非溶解),然后使其结晶化。通过适宜地选择熔解及结晶化的条件,从而可以得到dm为75~150μm、ca离子的含量低于1ppm的化合物(a)的粉末。制造方法及制造条件稍后详述。
[0093]
第1实施方式的粉末的na离子的含量低于1ppm,优选为0.7ppm以下,进一步优选为0.5ppm以下。通过除ca离子以外将na离子量也设为低于1ppm,从而能够将本实施方式的粉末更进一步适宜地用于电子设备制造。另外,na离子的含量也可以为零(装置的测定极限以下)。从实用性的观点出发,na离子的含量例如为0.01ppm以上。
[0094]
第1实施方式的粉末的mg离子的含量低于1ppm,优选为0.7ppm以下,进一步优选为0.5ppm以下。与ca离子同样,mg离子的含量也为计算水的硬度时可使用的指标,有时工业用水中会不可避免地含有mg离子。由此,除ca离子以外,mg离子量也设为低于1ppm,从而能够将本实施方式的粉末更进一步适宜地用于电子设备制造等各种用途。另外,mg离子的含量也可以为零(装置的测定极限以下)。从实用性的观点出发,mg离子的含量优选为例如0.01ppm以上。
[0095]
ca离子等金属离子的含量可通过利用离子色谱分析法求出。进行离子色谱分析时,通常制作使化合物(a)的粉末溶解于叔丁基甲醚等有机溶剂中而得的样品液体。对该样品液体中的金属离子的含量进行测定,由得到的测定值计算出“粉末中的ca离子的质量/粉末的质量”。
[0096]
在以往的bis-af的精制法、特别是使用水的再沉淀法中,由于所使用的水本身包含金属离子,因此大多数情况下金属离子量不易减少。如下述第1制法所述,使用水并通过
使化合物(a)“不积极地『溶解』”于水中的方法,而使化合物(a)重结晶,从而可以得到ca离子等金属离子量较少的粉末。
[0097]
第1实施方式的粉末优选不包含一元醇、二元醇等醇,或即便包含也为少量。更优选即便包含醇,也只是包含少量的水溶性的碳数4以下的一元醇,例如甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-甲基-1-丙醇、2-丁醇、2-甲基-2-丙醇等。
[0098]
第1实施方式的粉末中的醇的含量优选为400ppm以下,更优选为200ppm以下,进一步优选为100ppm以下。此处的“醇”不包括酚类化合物(具有酚性羟基的化合物)为化学常识,但为了慎重起见仍事先说明。
[0099]
由于粉末不包含醇,或即便包含也为少量,因此在使用本实施方式的粉末作为原料来制造聚合物时,不易发生意外反应,容易制造期望的聚合物。
[0100]
另外,由于化合物(a)与醇可以形成氢键,因此通过使粉末不包含醇、或即便包含也为少量,而具有容易准确称量纯化合物(a)的量的优点。该情况例如能够准确地控制例如使化合物(a)与环氧化合物反应时的“当量比”,而容易精密地控制最终得到的树脂的物性。
[0101]
顺便一提,在专利文献1中,在精制bis-af时使用了乙二醇,因此可认为在专利文献1记载的bis-af中包含超过400ppm的乙二醇。
[0102]
另一方面,在通过下述制造方法(第1制法)来制造第1实施方式的粉末时,有时使用醇作为使化合物(a)分散的分散介质的一部分。然而,由在第1制法不会使化合物(a)积极地“溶解”于醇中,因此粉末中的醇量较少。另外,在仅使用水作为分散介质的情况下,原理上化合物(a)不包含醇。
[0103]
化合物(a)中的醇量例如可利用气相色谱仪求出。
[0104]
在将第1实施方式的粉末的通过激光衍射散射法测得的体积基准累积50%直径设为d
50
时,d
50
优选为40~100μm,更优选为40~90μm,进一步优选为40~80μm。
[0105]
第1实施方式的粉末由于d
50
在特定数值范围内,因此操作性可进一步变好。由于d
50
相对较大,因此可认为颗粒彼此的接触面积变得更小,流动时的颗粒彼此的摩擦变得更小。此外,可认为致使过滤性、干燥性等进一步优化。
[0106]
在第1实施方式的粉末中,在将通过激光衍射散射法测得的体积基准累积90%直径设为d
90
时,(d
90-d
50
)/d
50
的值优选为1.3~1.7,更优选为1.4~1.7。
[0107]
(d
90-d
50
)/d
50
这一指标可以说是表示粒径分布曲线中大粒径侧的“下摆”的扩展状况的指标。该值为1.7以下是指至少大粒径侧的粒径分布相对尖锐。由于粒径分布尖锐,因此可认为粉末的均质性提高,操作性更进一步提高。
[0108]
另外,(d
90-d
50
)/d
50
为1.7以下的情况被认为也意味着粗大颗粒相对较少。由于粗大颗粒相对较少,例如也有可能可抑制块状化。
[0109]
顺便一提,作为(d
90-d
50
)/d
50
的优选范围的下限值的1.3是设定得到化合物(a)的粉末时的重结晶的成本及劳力不过度的范围者。
[0110]
作为与上述相似的指标,在第1实施方式的粉末中,(d
90-dm)/dm的值优选为0.93以下,更优选为0.92以下。(d
90-dm)/dm的值的下限例如为0.40以上。
[0111]
作为其它观点,在将第1实施方式的粉末的通过激光衍射散射法测得的平均粒径设为d
ave
时,d
ave
优选为45~80μm,更优选为45~60μm。作为较大的倾向,第1实施方式的粉末的平均粒径有大于以往的化合物(a)的粉末的倾向。
[0112]
在第1实施方式中,dm、d
50
等粒径的各种相关值可由通过激光衍射散射法测得的体积基准的粒径分布曲线求出。作为可进行利用激光衍射散射法的测定的装置,例如,可列举出岛津制作所株式会社的粒度分布计“sald”系列。测定通常是使粉末分散于实质上不溶解其的溶剂(例如正癸烷)中,进行湿式测定。
[0113]
(第2实施方式)
[0114]
在将第2实施方式的粉末的通过激光衍射散射法测得的体积基准累积50%直径设为d
50
、将通过激光衍射散射法测得的算术体积平均直径设为d
ave
时,d
50
为50~100μm,d
50
/d
ave
为1.1~1.5。
[0115]
第2实施方式的粉末的操作性良好的理由推测如下。
[0116]
若粉末包含的颗粒的粒径相对较大,则颗粒彼此的接触面积变得相对较小,因此,推测流动时的颗粒彼此的摩擦变小。
[0117]
在第2实施方式中,d
50
为50μm以上、即粉末中含有50%以上在体积基准中粒径为50μm以上的相对较大的颗粒,因此推测流动时的颗粒彼此的摩擦变小。
[0118]
另外,在第2实施方式中,d
50
/d
ave
为1.1以上表示纵轴绘制频度、横轴绘制粒径的粒径分布曲线的形状并非如图2所示左右对称(常态分布),而是如图1所示偏向右侧(粒径较大一侧)。即便是相同的d
50
,如图1的粒径分布中,相对较大的颗粒占全部颗粒的比例较大,因此颗粒彼此的接触面积容易变得更小。由此,推测流动时的颗粒彼此的摩擦变得更小。
[0119]
推测由此操作性良好。
[0120]
作为其它观点,第2实施方式的粉末的溶剂溶解性良好。即,意外的是,尽管本实施方式的粉末具有相对较大的d
50
,但却相对较快地溶于溶剂中。发明人等猜测其原因在于,由于粉末中小粒径的颗粒较少,因此溶剂中的bis-af的膨润及聚集得到抑制(若发生膨润及聚集,则粉末的分散性降低,溶解速度变慢)。
[0121]
顺便一提,作为d
50
的上限值的100μm是设定得到化合物(a)的粉末时的重结晶的成本及劳力不过度的范围者。d
50
/d
ave
的上限值1.5也同样如此。
[0122]
用于制造第2实施方式的粉末的方法及条件没有限定。然而,为了得到d
50
为50~100μm、d
50
/d
ave
为1.1~1.5的化合物(a)的粉末,优选为选择适当的方法及条件。
[0123]
在本实施方式中,例如在通过重结晶得到化合物(a)的粉末时,优选使用特定的有机溶剂、或使用晶种、或缓慢进行冷却。通过选择适当的制造方法及制造条件,可以得到d
50
为50~100μm、d
50
/d
ave
为1.1~1.5的化合物(a)的粉末。具体的制造方法后述作为“第2制法”说明。为了慎重起见事先说明,制造第2实施方式的粉末时,可以采用第1制法。
[0124]
如上所述,第2实施方式的粉末的d
50
为50~100μm,d
50
/d
ave
为1.1~1.5。
[0125]d50
优选为50~90μm,更优选为50~80μm,进一步优选为50~70μm。
[0126]d50
/d
ave
优选为1.1~1.4,更优选为1.1~1.3。
[0127]
在第2实施方式的粉末中,将通过激光衍射散射法测得的众数粒径(mode particle diameter)dm设于特定数值范围内,从而能够进一步改善流动性。由于dm相对较大,因此可认为颗粒彼此的接触面积变得更小,流动时的颗粒彼此的摩擦变得更小。此外,可认为操作性进一步提高。
[0128]
作为具体的数值,dm优选为75~150μm,更优选为80~120μm。
[0129]
第2实施方式的粉末中,在将通过激光衍射散射法测得的体积基准累积90%直径
设为d
90
时,(d
90-d
50
)/d
50
的值优选为1.3~1.7,更优选为1.4~1.7。
[0130]
(d
90-d
50
)/d
50
这一指标可以说是表示粒径分布曲线上右侧(大粒径侧)的“下摆”的扩展状况的指标。认为(d
90-d
50
)/d
50
为1.7以下是指大粒径侧的粒径分布相对尖锐,从与d
50
/d
ave
不同的观点出发,粉末的粒径分布是自如图1的常态分布偏离的分布。由此,由于除d
50
/d
ave
为1.1~1.5以外,(d
90-d
50
)/d
50
为1.7以下,因此可认为操作性进一步提高。
[0131]
另外,认为(d
90-d
50
)/d
50
为1.7以下也是指粗大颗粒相对较少。由于粗大颗粒相对较少,例如,也有可能可进一步抑制块状化。
[0132]
顺便一提,作为(d
90-d
50
)/d
50
的优选的范围的下限值的1.3是设定得到化合物(a)的粉末时的重结晶的成本及劳力不过度的范围者。
[0133]
在第2实施方式中,d
50
、d
90
、d
ave
及dm可由通过激光衍射散射法测得的体积基准的粒径分布曲线求出。作为可进行利用激光衍射散射法的测定的装置,例如,可列举出岛津制作所株式会社的粒度分布计“sald”系列。测定通常是使粉末分散于实质上不溶解其的溶剂(例如正癸烷),进行湿式测定。测定方法的详细内容请参照后述公开的实施例。
[0134]
(堆积密度)
[0135]
在第2实施方式的粉末的堆积密度在一定的数值范围内,因此可进一步提高粉末的操作性。
[0136]
具体而言,第2实施方式的粉末的松散堆积密度优选为0.50~0.75g/cm3,更优选为0.60~0.75g/cm3。另外,本实施方式的粉末的振实堆积密度优选为0.76~0.90g/cm3,更优选为0.80~0.90g/cm3。
[0137]
松散堆积密度及振实堆积密度的测定方法请参照后述公开的实施例。
[0138]
(第3实施方式)
[0139]
第3实施方式的粉末的d
50
为50~100μm。d
50
优选为50~90μm,更优选为50~80μm,进一步优选为50~70μm。
[0140]
在第3实施方式的粉末中,将通过激光衍射散射法测得的众数粒径(mode particle diameter)dm设在特定数值范围内,从而能够进一步改善流动性。由于dm相对较大,因此可认为颗粒彼此的接触面积变得更小,流动时的颗粒彼此的摩擦变得更小。此外,可认为操作性进一步提高。
[0141]
作为具体的数值,dm优选为75~150μm,更优选为80~120μm。
[0142]
第3实施方式的粉末中,在将通过激光衍射散射法测得的体积基准累积90%直径设为d
90
时,(d
90-d
50
)/d
50
的值优选为1.3~1.7,更优选为1.4~1.7。
[0143]
(d
90-d
50
)/d
50
这一指标可以说是表示粒径分布曲线上大粒径侧的“下摆”的扩展状况的指标。该值为1.7以下是指至少大粒径侧的粒径分布相对尖锐。由于粒径分布尖锐,因此可认为粉末的均质性提高,操作性更进一步提高。
[0144]
另外,(d
90-d
50
)/d
50
为1.7以下认为也是指粗大颗粒相对较少。由于粗大颗粒相对较少,例如,也有可能可进一步抑制块状化。
[0145]
顺便一提,作为(d
90-d
50
)/d
50
的优选的范围的下限值的1.3是设定得到化合物(a)的粉末时的重结晶的成本及劳力不过度的范围者。
[0146]
在第3实施方式中,d
50
、d
90
及dm可由通过激光衍射散射法测得的体积基准的粒径分布曲线求出。作为可进行利用激光衍射散射法的测定的装置,例如,可列举出岛津制作所株
式会社的粒度分布计“sald”系列。测定通常是使粉末分散于实质上不溶解其的溶剂(例如正癸烷),进行湿式测定。
[0147]
如上所述,第3实施方式的粉末的休止角为35~49
°
。休止角优选为40~49
°
,进一步优选为40~47
°
。
[0148]
休止角的测定方法请参照后述公开的实施例的记载。
[0149]
通过适当设计第3实施方式的粉末的堆积密度,可进一步提高粉末的操作性。
[0150]
具体而言,第3实施方式的粉末的松散堆积密度ρ1优选为0.50~0.75g/cm3,更优选为0.60~0.75g/cm3。另外,第3实施方式的粉末的振实堆积密度ρ2优选为0.76~0.90g/cm3,更优选为0.80~0.90g/cm3。
[0151]
松散堆积密度及振实堆积密度的测定方法请参照后述公开的实施例。
[0152]
特别是在第3实施方式中,ρ2/ρ1优选为1.01~1.45的范围内,更优选为1.10~1.40的范围内。通过使ρ2/ρ1为1.45以下,例如在称取本实施方式的粉末时能够充分减小称取质量的偏差。
[0153]
(第4实施方式)
[0154]
第4实施方式的粉末是2,2-双(4-羟基苯基)六氟丙烷(在通式(a)中r1~r8全部为氢原子者)的粉末,
[0155]
x射线衍射(xrd)光谱中的2θ=22.3
°
附近的峰的半值宽度为0.050
°
以上且0.180
°
以下,
[0156]
x射线衍射光谱中的2θ=23.7
°
附近的峰的半值宽度为0.050
°
以上且0.120
°
以下,
[0157]
x射线衍射光谱中的2θ=25.8
°
附近的峰的半值宽度为0.040
°
以上且0.120
°
以下。
[0158]
第4实施方式的粉末具体而言,具有“在溶剂、特别是极性溶剂或碱性溶剂中的溶解速度较快”、“可缩短对湿润的粉末进行干燥时的干燥时间”等优点。推测这些优点源于如下:x射线衍射光谱中的2θ=22.3
°
附近的峰的半值宽度为0.050
°
以上且0.180
°
以下,x射线衍射光谱中的2θ=23.7
°
附近的峰的半值宽度为0.050
°
以上且0.120
°
以下,x射线衍射光谱中的2θ=25.8
°
附近的峰的半值宽度为0.040
°
以上且0.120
°
以下。
[0159]
本发明人等着眼于粉末的x射线衍射光谱中特定的3个2θ处的峰的半值宽度。
[0160]
虽详情并不清楚,但可认为通过以2,2-双(4-羟基苯基)六氟丙烷所固有的2θ=22.3
°
附近的峰的半值宽度、2θ=23.7
°
附近的峰的半值宽度、及2θ=25.8
°
附近的峰的半值宽度变小的方式设计粉末,粉末的微晶的尺寸较大,且整个粉末的微晶的尺寸一致。
[0161]
由此,认为在溶解于溶剂中时,由微晶的尺寸不均所引起的聚集得到抑制,而在溶剂中的溶解速度变快。另外,在对湿润的粉末进行干燥时,因由微晶的尺寸不均所导致的聚集得到抑制,因此推测在粉末的颗粒之间不易存在液体成分,并且可缩短粉末的干燥时间。
[0162]
x射线衍射光谱中的2θ=22.3
°
附近是指以2θ=22.3
°
为中心
±1°
的范围,x射线衍射光谱中的2θ=22.3
°
附近的峰是指以2θ=22.3
°
为中心
±1°
的范围内的最大峰。
[0163]
x射线衍射光谱中的2θ=23.7
°
附近是指以2θ=23.7
°
为中心
±1°
的范围,x射线衍射光谱中的2θ=23.7
°
附近的峰是指以2θ=23.7
°
为中心
±1°
的范围内的最大峰。
[0164]
x射线衍射光谱中的2θ=25.8
°
附近是指以2θ=25.8
°
为中心
±1°
的范围,x射线衍射光谱中的2θ=25.8
°
附近的峰是指以2θ=25.8
°
为中心
±1°
的范围内的最大峰。
[0165]
2,2-双(4-羟基苯基)六氟丙烷的粉末的x射线衍射光谱中的2θ=22.3
°
附近的峰
的半值宽度为0.050
°
以上且0.180
°
以下。
[0166]
x射线衍射光谱中的2θ=22.3
°
附近的峰的半值宽度优选为0.055
°
以上,更优选为0.060
°
以上。
[0167]
另外,优选为0.170
°
以下,更优选为0.165
°
以下。
[0168]
2,2-双(4-羟基苯基)六氟丙烷的粉末的x射线衍射光谱中的2θ=23.7
°
附近的峰的半值宽度为0.050
°
以上且0.120
°
以下。
[0169]
x射线衍射光谱中的2θ=23.7
°
附近的峰的半值宽度优选为0.055
°
以上,更优选为0.060
°
以上。
[0170]
另外,优选为0.100
°
以下,更优选为0.090
°
以下。
[0171]
2,2-双(4-羟基苯基)六氟丙烷的粉末的x射线衍射光谱中的2θ=25.8
°
附近的峰的半值宽度为0.040
°
以上且0.120
°
以下。
[0172]
x射线衍射光谱中的2θ=25.8
°
附近的峰的半值宽度优选为0.045
°
以上,更优选为0.050
°
以上。
[0173]
另外,优选为0.115
°
以下,更优选为0.110
°
以下。
[0174]
粉末的x射线衍射(xrd)光谱可以通过利用辐射照射的x射线粉末衍射计(smart lab;rigaku公司制)进行测定。
[0175]
测定样品是将试样在研钵中研碎后,采集至在测定范围内不具有衍射峰的无反射试样板(可以自rigaku公司等购入),使其平坦化,将各试样板置于装置进行测定。测定条件的一例如下。
[0176]
[x射线衍射的测定条件]
[0177]
球管:cu
[0178]
电压:40kv
[0179]
电流:50ma
[0180]
太阳能缝隙:2.5
°
(入射侧、受光侧)
[0181]
扫描范围:10~80
°
[0182]
步宽:0.01
°
[0183]
扫描速度:35
°
/分钟
[0184]
检测器:一维x射线检测器(d/tex ultra250;rigaku公司制)
[0185]
在由测定得到的x射线衍射光谱中,分别算出2θ=22.3
°
附近、23.7
°
附近、25.8
°
附近的峰的半值宽度(θ为布勒格角)。峰的半值宽度的算出可利用smart lab studioii(解析软件:power xrd)进行。
[0186]
用于制造第4实施方式的粉末的方法及条件没有限定。例如,如下述第1制法所说明,使原料物质在水系分散介质中熔融,然后以下述结晶化工序的温度t2维持充分的时间(例如,30分钟以上),使其结晶化,由此,可较佳地得到x射线衍射(xrd)光谱中的2θ=22.3
°
附近的峰、2θ=23.7
°
附近的峰、及2θ=25.8
°
附近的峰的半值宽度分别在上述范围内的粉末。
[0187]
第4实施方式的粉末的粒径没有特别限定,具有适度的操作性即可。例如,可将通过激光衍射散射法测得的体积基准累积50%直径(d
50
)设为优选为20~100μm。另外,也可将下限设为更优选为30μm以上、进一步优选为40μm以上,可以将上限设为更优选为90μm以下、
进一步优选为80μm以下。
[0188]
作为可进行利用激光衍射散射法的测定的装置,例如,可列举出岛津制作所株式会社的粒度分布计“sald”系列。测定通常是使粉末分散于实质上不溶解其的溶剂(例如正癸烷)中,进行湿式测定。
[0189]
《粉末的制造方法》
[0190]
第1~第4实施方式的粉末可通过适当的制造方法而制造。以下,对用于制造第1~第4实施方式的粉末的优选的两个制造方法(第1制法及第2制法)进行说明。
[0191]
(第1制法)
[0192]
通式(a)所示的化合物的粉末的制造方法(第1制法)例如
[0193]
可包括:
[0194]
·
熔解工序,通过将包含通式(a)所示的化合物的原料物质、及水系分散介质放入容器中并进行加热,从而使原料物质在水系分散介质的存在下熔解而得到包含原料物质的熔解物及水系分散介质的不均匀液体;
[0195]
·
结晶化步骤,即通过使不均匀液体降温,而使熔解物结晶化,从而得到结晶。
[0196]
在第1制法的熔解工序中,原料物质的熔解温度t1下的通式(a)所示的化合物于水系分散介质中的溶解度为10[g/100g]以下,优选为8[g/100g]以下。该溶解度的下限值可为零,通常溶解度为0.5[g/100g]以上。
[0197]“原料物质的熔解温度t
1”是指粉末状(固体状)的原料物质全部熔解、失去原形的最低温度。原料物质全部熔解可通过以下方法等进行确认:(i)对容器内的原料物质的状态进行目视观察,确认不存在粉末状(固体状)的原料物质,由此确认原料物质全部熔解;(ii)对自容器内抽出的水层迅速进行目视观察,确认原料物质不以粉末状(固体状)的形态残留。
[0198]
对于第1制法,也可如下说明。
[0199]
·
于熔解工序中,将包含化合物(a)的原料物质在水系分散介质中加热,在温度t1下使其熔解(非溶解),而制成熔解物。水系分散介质简单而言,在温度t1下为化合物(a)的“不良溶剂”(不易使化合物(a)溶解的液体)。通过于化合物(a)的不良溶剂(水系)中加热化合物(a),得到包含原料物质的熔解物及水系分散介质的不均匀液体。
[0200]
·
在结晶化步骤中,使上述不均匀液体中的熔解物结晶化。
[0201]
在第1制法中,于原料物质的熔解物与水系分散液的界面,ca离子等自原料物质侧向水系分散液侧移动,因此可认为能够减少ca离子等的量。从制造第1实施方式的粉末(ca离子较少)的观点出发,是优选的。
[0202]
另外,在第1制法中,由于不使原料物质积极地“溶解”(不经过溶质与溶剂完全均匀混合的状态),因此可认为可抑制作为源自水等溶剂的杂质的ca离子等被混入原料物质经重结晶者。同样地,认为暂时自原料物质溶出的杂质即ca离子等“再次”被并入已结晶化的原料物质会得到抑制。认为这些也关系到能够减少ca离子等杂质的量。
[0203]
顺便一提,例如bis-af即便其单独加热至100℃(水的沸点)也不熔解,然而,水中的bis-af加热至100℃(或低于100℃的温度)便熔解。根据公知信息及本发明人等的见解,其原因在于,bis-af通过水和而熔点下降。公知信息请参照例如上述专利文献3。
[0204]
第1制法也具有环境负荷较小的额外的优点。具体而言,实施第1制法时,主要使用
水系分散液,不需要有机溶剂(化合物(a)的良溶剂)。即,通过采用上述制造方法,能够减少有机溶剂的使用,因此,可减小环境负荷。
[0205]
以下,对熔解工序及结晶化工序详细进行说明。
[0206]
·
熔解工序
[0207]
通常在熔解工序中,将包含化合物(a)的原料物质、及水系分散介质在具备搅拌机构及加热机构的容器内加热。如此,可以得到包含原料物质的熔解物及水系分散介质的不均匀液体。通过一边搅拌一边加热,不均匀液体通常变更为悬浮状态的液体。在不进行搅拌或停止搅拌并静置的情况下,不均匀液体通常为2层分离状态。顺便一提,通过一边搅拌一边加热,认为原料物质的熔解物与水系分散介质充分地接触,ca离子等更容易自原料物质侧向水系分散液侧移动。
[0208]
作为一例,水系分散液可实质上仅包含水。通过使用实质上仅包含水的水系分散液,可使最终得到的化合物(a)中的醇量实质上为零。另外,由于化合物(a)在水中的溶解性非常小,因此也具有能够减少废液中所含的化合物(a)的优点。
[0209]
作为另一例,水系分散液可包含水及醇。在该情况,水系分散介质中的醇的比率通常为30质量%以下,优选为1~30质量%,进一步优选为5~25质量%。若醇的比率过多,则有可能原料物质不会适宜分散于水系分散液中,其大部分“溶解”。由此,水系分散液中的醇的比率优选设为上述程度。
[0210]
通过使用包含水及醇者作为水系分散液,具有可降低温度t1的优点。根据本发明人等的见解,在仅使用水作为水系分散液的情况下,t1为90℃以上,而通过水系分散液包含30质量%以下的醇,有t1低于90℃(为30~90℃左右)的倾向。由于t1变低,因此容易进行微妙的温度调整,例如,在后述的结晶化工序中容易使结晶生长。
[0211]
关于原料物质与水系分散液的比率,可考虑ca离子等杂质的减少效果(使用的水系分散液越多,则原料物质中的ca离子等越容易向水系分散液的侧移动)、与成本的兼顾而适当设定。水系分散液相对于原料物质100质量份的量通常为100~3000质量份,优选为200~1500质量份。
[0212]
温度t1可根据化合物(a)的具体结构及水系分散液的组成而变化。在熔解工序中根据所需的加热温度及时间进行适宜设定。在熔解工序中,全部化合物(a)中,除溶解于水系分散液中的量以外全部熔解即可。
[0213]
·
结晶化工序
[0214]
在结晶化工序中,通过使熔解工序中得到的不均匀液体降温,从而使不均匀液体中的熔解物结晶化。通过适宜地选择降温的具体条件,可以得到ca离子等较少、且粒径分布得到适当控制(例如第1实施方式所述,dm为75~150μm)的化合物(a)的粉末。另外,通过适宜地选择降温的具体条件,或许可进行微晶的尺寸的控制等,而可以得到xrd光谱的半值宽度相对较小的化合物(a)的粉末。
[0215]
第1制法中的结晶化工序与一般的结晶化工序、即使溶解于均匀的“溶液”的物质结晶化的工序不同。
[0216]
在一般的结晶化工序中,随着结晶析出,需要缓慢降低溶液的温度。
[0217]
另一方面,第1制法中的结晶化工序将不均匀液体降温至一定温度以下(低于熔解温度)即可,未必需要“缓慢降低”温度。由于大部分化合物(a)仅“熔解”而不“溶解”,因此只
要将温度维持在一定温度以下(低于熔解温度),大部分化合物(a)便可结晶化(固化)。通过这样的不同寻常的结晶化机理,得到的结晶(最终物)与以往的结晶不同。
[0218]
其中,在例如水系分散介质包含醇的情况下,由于一定程度的量的化合物(a)“溶解”于分散介质中,因此可以以适当的速度进行降温操作。另外,在结晶化工序中,可在维持低于熔解温度的一定温度后,进行降温操作。
[0219]
结晶化工序的温度t2优选为比温度t1低1~10℃,更优选为低1~8℃。具体而言,在结晶化工序中,优选为将体系在上述温度t2(比温度t1低1~10℃的范围内)维持优选为30分钟以上、更优选为60分钟以上。通过将温度t2维持充分长的时间,容易得到ca离子等充分减少、且粒径分布得到适当控制(例如第1实施方式所述,dm为75~150μm)的结晶。另一方面,从制造的效率性的观点出发,在结晶化工序中维持温度t2的时间优选为例如300分钟以下。
[0220]
结晶化工序优选一边搅拌不均匀液体一边来进行。在熔解工序中进行搅拌的情况下,优选直接继续搅拌。如此,容易充分地减少ca离子等杂质的量。另外,由于分散液经常搅拌,因此容易得到结晶生长得到适当控制、粒径分布得到适当控制(例如第1实施方式所述,dm为75~150μm)的粉末。上述搅拌优选为以50~500rpm的速度进行,更优选为以150~300rpm的速度进行。
[0221]
在结晶化工序中,可使用晶种,也可不使用晶种。在使用晶种的情况下,其添加量以质量比计可设为分散于水系分散介质的化合物(a)的1/1000至1/100左右。晶种没有特别限定,为固体状的化合物(a)即可。
[0222]
·
结晶化工序后的处理
[0223]
在结晶化工序后,将体系通常冷却至常温(25℃左右)。
[0224]
冷却的条件没有特别限定。可为自然冷却,也可以不是自然冷却。如上所述,本实施方式是主旨不同于使“溶解”的化合物(a)结晶化的现有技术者,因此,大量化合物(a)不通过冷却而析出。
[0225]
然而,由于水系分散介质使少量化合物(a)溶解,因此少量结晶通过冷却而析出。从ca离子等的量及粒径分布的精密调整的观点出发,冷却优选为以0.1~0.3[℃/分钟]左右缓慢进行。
[0226]
冷却后,例如通过减压过滤回收得到的结晶,用水清洗,在20~50℃左右的环境下进行减压干燥,从而可以得到最终的粉末。
[0227]
(第2制法)
[0228]
通式(a)所示的化合物的粉末的制造方法(第2制法)包括例如以下的工序1~工序4的一系列工序。通过这样的工序,可以得到粒径分布得到适当控制(例如第2实施方式所述,d
50
为50~100μm,d
50
/d
ave
为1.1~1.5)的化合物(a)的粉末。
[0229]
·
工序1:原料的准备
[0230]
首先,准备包含通式(a)所示的化学结构的物质作为原料。这样的原料例如可通过上述专利文献2中记载的方法而得到。原料可使用市售品。
[0231]
·
步骤2:在有机溶剂中的溶解(加热等)
[0232]
将工序1中准备的原料投入有机溶剂中。此外,一边搅拌有机溶剂,一边加热,从而使原料溶解于有机溶剂中。
[0233]
此时,重要的是选择适当的溶剂作为有机溶剂。具体而言,优选使用包含化合物
(a)的不良溶剂及良溶剂的混合溶剂作为有机溶剂。由此,在后述的(3)重结晶中,结晶容易缓慢生长,容易得到粒径分布得到适当控制(例如第2实施方式所述,d
50
及d
50
/d
ave
得到适当控制)的粉末。
[0234]
作为不良溶剂,例如,可列举出:环己烷、甲基环己烷、乙基环己烷等脂环族烃系溶剂;正戊烷、正己烷、异己烷、正庚烷、正辛烷、异辛烷、正癸烷等脂肪族烃系溶剂。
[0235]
作为良溶剂,可列举出乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸丁酯、乙酸异丁酯、乙酸戊酯、乙酸异戊酯、乳酸甲酯、乳酸乙酯、乳酸丁酯等酯系溶剂。
[0236]
不良溶剂与良溶剂的混合比以质量比计,例如为不良溶剂/良溶剂=99/1~50/50,优选为不良溶剂/良溶剂=95/5~75/25,更优选为不良溶剂/良溶剂=95/5~80/20。
[0237]
在工序2中,有机溶剂例如加热至60~80℃左右。通过该加热及搅拌,使原料完全溶解于有机溶剂中。换言之,对于向有机溶剂中的原料的投入量,在将有机溶剂加热至60~80℃左右时完全溶解,以在下述工序3(降温)中发生析出的程度的量加以调整。该量可基于原料的溶解性、使用的有机溶剂、加热温度等而适当设定。
[0238]
原料的用量(向有机溶剂中的投入量)可设为每1000g有机溶剂为100g左右,但该用量仅是一个例子。
[0239]
·
步骤3:降温/晶种的添加/搅拌
[0240]
在工序2中,将加热至60~80℃左右从而原料完全溶解的有机溶剂,用30分钟~3小时左右缓慢降温至55℃左右。
[0241]
降温后,在有机溶剂中添加固体状的化合物(a)作为晶种。根据本发明人等的见解,特别是使用如上所述的不良溶剂及良溶剂作为有机溶剂的情况下,通过使用晶种,最终容易得到粒径分布得到适当控制(例如第2实施方式所述,d
50
及d
50
/d
ave
被控制)的化合物(a)的粉末。
[0242]
晶种的添加量以质量比计,可设为工序2中溶解于有机溶剂的原料的1/1000至1/100左右。晶种没有特别限定,为固体状的化合物(a)即可。例如可使用依据上述的专利文献2的实施例中记载的方法所得者作为晶种。另外,也可使用市售的固体状的化合物(a)作为晶种。
[0243]
添加晶种后,将温度维持在55℃左右,在该状态下将有机溶剂搅拌30分钟~3小时左右,使结晶析出。
[0244]
·
工序4:冷却
[0245]
在工序3后,一边搅拌有机溶剂,一边用约1~5小时缓慢冷却,直至温度变为30~40℃左右。由此,使结晶生长。
[0246]
通过减压过滤回收得到的结晶,并在20~50℃左右的环境下进行减压干燥。如此,可以得到粒径分布等得到适当控制的粉末。
[0247]
《溶液的制造方法》
[0248]
利用溶剂、及第1~第4实施方式的至少任一实施方式的粉末,可制造包含通式(a)所示的化合物的溶液。通过使用该溶液,可制造各种低分子化合物、低聚物、聚合物等。
[0249]
在制造溶液时,(i)可将第1~第4实施方式的至少任一实施方式的粉末直接添加至溶剂中,进行搅拌或使其溶解,(ii)也可首先对第1~第4实施方式的至少任一实施方式的粉末进行微细化而得到微细化的粉末,将该至少微细化的粉末添加至溶剂中进行搅拌或
使其溶解。
[0250]
如上所述,第1~第4实施方式的粉末在工业上的操作性良好。例如在第1实施方式中,由于众数粒径dm为75~150μm,因此具有“过滤性良好”、“可缩短对湿润的粉末进行干燥时的干燥时间”等优点。另一方面,在众数粒径dm相对较大为75~150μm的情况下,也存在根据溶剂的种类溶解需要一点时间的顾虑。由此,有时优选如上述(ii)所述制造溶液的情况。
[0251]“微细化”的方法没有特别限定。代表性的微细化的方法为粉碎。工业上而言,可利用喷射式粉碎机、辊磨机、锤磨机、针磨机、旋转研磨机、振动磨机、行星球磨机、珠磨机等装置进行粉碎。
[0252]
作为微细化的另一方法,可将第1~第4实施方式的至少任一实施方式的粉末投入溶剂中,在完全溶解前(例如通过过滤)取出未溶解的粉末,而得到微细的粉末。
[0253]
作为微细化的另一方法,可将第1~第4实施方式的至少任一实施方式的粉末投入全部粉末不会溶解的量的溶剂中,粉末的溶解饱和后,取出未溶解的粉末,由此得到微细的粉末。或者,也可直接使用存在未溶解的粉末的溶液。
[0254]
在进行微细化的情况下,以何种程度进行微细化没有特别限定。可根据微细化期望的成本、进行微细化的优点(例如上述的溶解性的提高)的兼顾,决定以何种程度进行微细化。作为一个观点,以在微细化(优选为粉碎)的前后、d
ave
为1/20~1/2、更优选为1/15~1/4的方式进行微细化即可。从d
ave
的绝对值的观点出发,以微细化(优选为粉碎)后的粉末的d
ave
优选为0.1~15μm、更优选为0.5~10μm的方式进行微细化即可。
[0255]
制造化合物(a)的溶液时可使用的溶剂可根据各种目的而适宜地选择。可使用的溶剂并无特别限制,使化合物(a)溶解即可。具体而言,可示例:n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基甲酰胺、六甲基磷酸三酰胺、n-甲基-2-吡咯啶酮等酰胺系溶剂;甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-甲基-1-丙醇、2-丁醇、2-甲基-2-丙醇等醇系溶剂;二乙醚、二丙醚、二异丙醚、二丁醚、环戊基甲醚、二苯醚、二甲氧基乙烷、二乙氧基乙烷、四氢呋喃、二氧杂环己烷1-甲氧基-2-丙醇等醚系溶剂;乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙二醇单甲醚乙酸酯、乳酸乙酯等酯系溶剂;乙腈、丙腈、苯甲腈等腈系溶剂;苯、甲苯、二甲苯、乙苯、异丙苯等芳香族烃系溶剂;二氯甲烷、氯仿、1,2-二氯乙烷、1,1,2,2-四氯乙烷等卤素系溶剂;γ-丁内酯、γ-戊内酯、δ-戊内酯、ε-己内酯的内酯系溶剂。溶剂可单独使用,也可两种以上组合使用。
[0256]
以上,对本发明的实施方式进行了说明,然而,这些只是本发明的示例,也可以采用上述以外的各种构成。另外,本发明并不限定在上述实施方式,本发明包括可实现本发明的目的的范围内的变化、改良等。
[0257]
[实施例]
[0258]
基于实施例及比较例,对本发明的实施方式详细进行说明。为了慎重起见事先说明,本发明并不仅限定在实施例。
[0259]
[实施例1~5及比较例1~6]
[0260]
以下的实施例1~5及比较例1~6特别是用于对第1实施方式详细进行说明的实施例及比较例。
[0261]
《实施例1》
[0262]
自试剂瓶采集central glass公司制造的bis-af粉末25.0g(74.4mmol),并将其置
于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水225.0g。此外,一边搅拌一边将容器内部的温度升温至95℃。
[0263]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下将温度维持在95℃并搅拌1小时。
[0264]
经过1小时后,暂时停止搅拌并静置,对容器内进行观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0265]
此时,对水层侧的bis-af浓度进行测定。浓度为1质量%左右,尽管为95℃左右的高温,大部分bis-af也未“溶解”于水中。即,在原料的bis-af粉末的熔解温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0266]
接着,使内温自95℃略微降低,一边维持92~94℃的温度一边搅拌1小时。如此,结晶析出。然后,将内温以15℃/1小时的降温速度冷却至室温。此时的室温约为25℃。
[0267]
冷却后,针对析出的bis-af,使用具备滤纸的吸滤器,通过减压过滤进行分离并回收。回收后用75ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在75℃、减压(1kpa以下)下干燥8小时。
[0268]
干燥后得到的bis-af粉末为24.1g,收率为96%。
[0269]
《实施例2》
[0270]
自与实施例1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水202.5g及甲醇22.5g,一边搅拌一边将容器内部的温度升温至66℃。
[0271]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下维持温度并搅拌1.5小时。
[0272]
经过1.5小时后,暂时停止搅拌并静置,对容器内进行观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0273]
此时,对水层侧的bis-af浓度进行测定。浓度为2质量%以下,尽管为66℃左右的高温,大部分bis-af也未“溶解”于水/甲醇中。即,在原料的bis-af粉末会完全“熔解”的温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0274]
接着,一边将内温维持在60~65℃的温度一边搅拌1小时。如此,结晶析出。然后,将内温以15℃/1小时的降温速度冷却至室温。需要说明的是,此时的室温约为25℃。
[0275]
冷却后,针对析出的bis-af,使用具备滤纸的吸滤器,通过减压过滤进行分离并回收。回收后用75ml的纯水进行挂洗。使用真空干燥器,将清洗过的bis-af在80℃、减压(1kpa以下)下干燥6小时。
[0276]
干燥后得到的bis-af粉末为22.9g,收率为92%。
[0277]
《实施例3》
[0278]
自与实施例1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水112.5g及甲醇12.5g,一边搅拌一边将容器内部的温度升温至66℃。
[0279]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下维持温度,
并搅拌2小时。
[0280]
经过2小时后,暂时停止搅拌并静置,对容器内进行观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0281]
此时,对水层侧的bis-af浓度进行测定。浓度为2质量%以下,尽管为66℃左右的高温,大部分bis-af也未“溶解”于水/甲醇中。即,在原料的bis-af粉末会完全“熔解”的温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0282]
接着,一边将内温维持在59~65℃的温度一边搅拌1小时。如此,结晶析出。然后,将内温以15℃/1小时的降温速度冷却至室温。需要说明的是,此时的室温约为25℃。
[0283]
冷却后,针对析出的bis-af,使用具备滤纸的吸滤器,通过减压过滤进行分离并回收。回收后用75ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在80℃、减压(1kpa以下)下干燥6小时。
[0284]
干燥后得到的bis-af粉末为23.4g,收率为94%。
[0285]
《实施例4》
[0286]
自与实施例1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水100.0g及甲醇25.0g,一边搅拌一边将容器内部的温度升温至40℃。
[0287]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下维持温度,并搅拌2小时。
[0288]
经过2小时后,暂时停止搅拌并静置,对容器内进行观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0289]
此时,对水层侧的bis-af浓度进行测定。浓度为2质量%以下,尽管为40℃左右的温度,大部分bis-af也未“溶解”于水/甲醇中。即,在原料的bis-af粉末会完全“熔解”的温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0290]
接着,一边将内温维持在34~39℃的温度一边搅拌1小时。如此,结晶析出。然后,将内温以10℃/1小时的降温速度冷却至室温。需要说明的是,此时的室温约为25℃。
[0291]
冷却后,针对析出的bis-af,使用具备滤纸的吸滤器,通过减压过滤进行分离并回收。回收后用75ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在80℃、减压(1kpa以下)下干燥6小时。
[0292]
干燥后得到的bis-af粉末为23.0g,收率为92%。
[0293]
《实施例5》
[0294]
自试剂瓶采集central glass公司制造的bis-af粉末100.0g(298mmol),并将其置于具备搅拌机及冷却冷凝器的容积为2000ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水425.0g及甲醇75.0g。此外,一边搅拌一边将容器内部的温度升温至55℃。
[0295]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下将温度维持在55℃并搅拌1小时。
[0296]
经过1小时后,暂时停止搅拌并静置,对容器内进行观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解
的bis-af。
[0297]
此时,对水层侧的bis-af浓度进行测定。浓度为1质量%左右,大部分bis-af未“溶解”于水中。即,在原料的bis-af粉末的熔解温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0298]
接着,使内温自55℃略微降低,一边维持在49~50℃的温度一边搅拌1小时。如此,结晶析出。然后,将内温以15℃/1小时的降温速度冷却至室温。此时的室温约为25℃。
[0299]
冷却后,使用具备滤纸的吸滤器,通过减压过滤分离并回收析出的bis-af。回收后用300ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在75℃、减压(1kpa以下)下干燥8小时。
[0300]
干燥后得到的bis-af粉末为95.1g,收率为95%。
[0301]
《比较例1》
[0302]
比较例1是尝试单纯利用水清洗而减少ca离子的例子。
[0303]
自与实施例1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机的容积为300ml的硼硅酸玻璃制容器中。接着,添加纯水125.0g,一边于室温(约20℃)下搅拌1小时,一边清洗bis-af粉末。
[0304]
清洗结束后,使用具备滤纸的吸滤器,通过减压过滤进行分离并回收。
[0305]
接着,使用真空干燥器,在80℃、减压(1kpa以下)下干燥6小时。
[0306]
干燥后得到的bis-af粉末为24.0g,收率为96%。
[0307]
《比较例2》
[0308]
比较例2是为了减少结晶性化合物的金属量而屡次尝试的、尝试利用再沉淀而减少ca离子的例子。
[0309]
将纯水200.0g置于具备搅拌机的容积为300ml的硼硅酸玻璃制容器中。
[0310]
接着,一边转动搅拌机,一边将使自与实施例1相同的试剂瓶采集的bis-af粉末25.0g(74.4mmol)溶解于甲醇50.0g中得到的溶液、缓慢滴加于其内有上述纯水的硼硅酸玻璃制容器中。由此,使bis-af的固体析出。
[0311]
使用具备滤纸的吸滤器,通过减压过滤回收析出的bis-af。接着,使用真空干燥器,在80℃、减压(1kpa以下)下干燥8小时。
[0312]
干燥后得到的bis-af粉末为23.2g,收率为93%。
[0313]
《比较例3》
[0314]
比较例3是参考专利文献(cn104529717a)的记载、尝试利用活性碳减少ca离子的例子。
[0315]
自与实施例1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机的容积为300ml的硼硅酸玻璃制容器中。接着,在容器中添加甲醇50.0g、活性碳(osaka gas chemical株式会社制,商品名shirasagi anox-1)2.0g,在室温(约20℃)下搅拌5小时。
[0316]
搅拌结束后,使用具备滤纸的吸滤器通过过滤去除活性碳,回收bis-af的甲醇溶液。对甲醇溶液的金属离子浓度进行测定,其结果,na离子为9ppm,ca离子为12ppm。
[0317]
接着,将该甲醇溶液缓慢滴加于纯水200g中,由此,使bis-af再沉淀。bis-af析出后,使用具备滤纸的吸滤器,通过减压过滤回收bis-af。接着,使用真空干燥器,在80℃、减压(1kpa以下)下干燥8小时。
[0318]
干燥后得到的bis-af粉末为21.9g,收率为88%。
[0319]
《比较例4》
[0320]
比较例4是将比较例3中的活性碳替换成离子交换树脂得到的例子。
[0321]
自与实施例1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机的容积为300ml的硼硅酸玻璃制容器中。接着,在容器中添加甲醇50.0g、离子交换树脂(sumika chemtex株式会社制,商品名duolite c255lfh)2.0g,在室温(约20℃)下搅拌5小时。
[0322]
搅拌结束后,使用具备滤纸的吸滤器通过过滤去除离子交换树脂,回收bis-af的甲醇溶液。对甲醇溶液的金属离子浓度进行测定,其结果,na离子为3ppm,ca离子为2ppm。
[0323]
接着,将该甲醇溶液缓慢滴加于纯水200g中,由此,使bis-af再沉淀。bis-af析出后,使用具备滤纸的吸滤器,通过减压过滤回收bis-af。接着,使用真空干燥器,在80℃、减压(1kpa以下)下干燥7小时。
[0324]
干燥后得到的bis-af粉末为22.2g,收率为89%。
[0325]
《比较例5》
[0326]
比较例5是将比较例3中的活性碳替换成活性白土的例子。
[0327]
自与实施例1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机的容积为300ml的硼硅酸玻璃制容器中。接着,在容器中添加甲醇50.0g、活性白土(水泽化学工业株式会社制,商品名galleon earth)2.0g,在室温(约20℃)下搅拌5小时。
[0328]
搅拌结束后,使用具备滤纸的吸滤器通过过滤去除活性白土,回收bis-af的甲醇溶液。对甲醇溶液的金属离子浓度进行测定,其结果,na离子为18ppm,ca离子为4ppm。
[0329]
(由于该甲醇溶液中的金属浓度明显高于比较例4,因此推测即便进行之后的再沉淀,恐怕也无法去除金属,因此未进行再沉淀)。
[0330]
《比较例6》
[0331]
比较例6是参照日本特开2007-246819号公报中记载的事项得到的例子。
[0332]
在比较例6中,使用水与醇(乙二醇)的混合溶剂。然而,从在一系列流程中原料的bis-af粉末完全溶解于混合溶剂(成为均匀的1层)的方面而言,比较例6与本实施方式本质上不同。
[0333]
自与实施例1相同的试剂瓶将bis-af粉末150g置于具备搅拌机及冷却冷凝器的容积为2l的硼硅酸玻璃制容器中,向其中注入乙二醇300g及离子交换水700g。然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为85℃,一边使bis-af溶解。在该状态下维持温度,并搅拌1小时。经过1小时后,暂时停止搅拌并静置,对容器内进行观察。如此,确认溶液均匀。
[0334]
接着,一边继续搅拌,一边以10℃/小时的降温速度进行冷却直至烧瓶的内温变更为25℃。如此,开始降温的同时,结晶开始缓慢析出(再结晶)。通过减压(1kpa以下)过滤回收析出的结晶,并在60℃下进行减压干燥。
[0335]
干燥后得到的bis-af粉末为137.0g,收率为91%。此外,na离子的浓度为0.2ppm,ca离子的浓度为0.3ppm。
[0336]
《测定》
[0337]
(粒径分布)
[0338]
利用岛津制作所制造的粒度分布计sald-2200,对使得到的粉末状bis-af分散于正癸烷溶剂中得到的样品的粒径分布进行测定。测定时,预先在载玻片上将bis-af与正癸烷溶剂混合,准备了糊剂状的bis-af。然后,向作为分散溶剂的正癸烷中一点点地添加糊剂状的bis-af,以吸光度的数值成为0.1l/(mol
·
cm)以下的方式调整添加量,对粒度分布进行测定。
[0339]
通过对得到的测定结果(粒度分布)进行解析,计算出dm等。
[0340]
(ca、na及mg离子的含量)
[0341]
利用离子色谱分析法,求出粉末中的ca、na及mg离子的含量。分析法详细如下。
[0342]
首先,将实施例/比较例中得到的各bia-af粉末0.4g溶解于叔丁基甲醚2.0ml,制成溶液。将该溶液及超纯水2.0ml置于分液漏斗中,剧烈振荡分液漏斗,由此,在超纯水(水层)一侧提取金属离子成分。将分液漏斗静置,将分离的水层的侧作为金属离子成分的分析用的分析样品液体。
[0343]
利用thermo fisher scientific制造的离子色谱法装置(cs-2100),对该分析样品液体的金属离子含量进行测定。作为柱,使用分离柱(ion pac cs16(内径4mm
×
250mm))及保护柱(ion pac cg16(内径4mm
×
100mm))。另外,作为洗脱液,使用30mm甲磺酸,洗脱液流量为1.0ml/分钟,温度设为35℃。
[0344]
将粒径分布、及ca、na及mg离子的含量的相关信息汇总示于下表。顺便一提,在一部分比较例中,由于金属离子的含量明显较大,因此未测定粒径分布(表中用n.d.表示)。
[0345]
[表1]
[0346][0347]
(醇的含量)
[0348]
在实施例2中,将干燥后的bis-af 1.00g溶解于1.00g的乙酸乙酯中,进行气相色谱分析。在以除乙酸乙酯以外的峰面积为基准时,甲醇含量低于100ppm。
[0349]
在比较例6中,将干燥后的bis-af 1.00g溶解于1.00g的乙酸乙酯中,进行气相色谱分析。在以除乙酸乙酯以外的峰面积为基准时,乙二醇含量为500ppm。
[0350]
《评价》
[0351]
使bis-af粉末分散于水中的分散液通过具备滤纸的吸滤器,从而将粉末与水分
离。如下对此时的过滤性(脱液)的良好性进行评价。
[0352]
·
良好:减压开始后,自过滤器(漏斗底部)的滴液迅速消失。
[0353]
·
差:减压开始后,自过滤器(漏斗底部)的滴液持续片刻。
[0354]
另外,通过卡尔费休法,对进行抽吸过滤直至自过滤器(漏斗底部)的滴液消失后的粉末的含水率进行测定。
[0355]
将评价结果汇总示于下表。顺便一提,如上所述,由于比较例5中未进行再沉淀,因此也不存在脱液及含水率的评价结果(n.d.=no data)。
[0356]
[表2]
[0357]
表2
[0358][0359]
如表2所示,实施例1~5的bis-af的粉末的过滤性良好。另外,自含水率的评价结果可知,对实施例1~5的bis-af的粉末进行干燥时,干燥时间较短即可。即,实施例1~5的bis-af的粉末在工业上的操作性优异。
[0360]
此外,实施例1~5的bis-af的粉末的ca离子的含量低于1ppm。由此可知,实施例1~5的bis-af的粉末可适宜用于各种技术领域(例如电子设备制)。
[0361]
[实施例2-1~2-4及比较例2-1~2-3]
[0362]
以下的实施例2-1~2-4及比较例2-1~2-3特别是用于对第2实施方式详细进行说明的实施例及比较例。
[0363]
《实施例2-1》
[0364]
首先,将bis-af(central glass公司制)200g置于具备搅拌马达、温度计及冷却冷凝器的硼硅酸玻璃制的3l烧瓶内,向其中注入正己烷1800g及乙酸乙酯200g。
[0365]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0366]
在烧瓶的内温达到65℃后,用1小时缓慢降温直至内温变为55℃,在该降温后,添加bis-af(central glass公司制)1g作为晶种,在该状态下搅拌1小时,使目标bis-af析出。
[0367]
进而,然后,冷却2小时直至内温变为35℃,通过减压过滤回收析出的结晶。在30℃下对回收的结晶进行减压干燥,由此,得到24g的粉末状bis-af。
[0368]
《实施例2-2》
[0369]
首先,将bis-af(central glass公司制)200g置于具备搅拌马达、温度计及冷却冷凝器的硼硅酸玻璃制的3l烧瓶内,向其中注入正己烷1800g及乙酸乙酯200g。
[0370]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0371]
在烧瓶的内温达到65℃后,用2小时缓慢降温直至内温变为55℃,在该降温后,添加bis-af(central glass公司制)1g作为晶种,在该状态下搅拌2小时,使目标bis-af析出。
[0372]
进而,然后,冷却4小时直至内温变为35℃,通过减压过滤回收析出的结晶。在30℃下对回收的结晶进行减压干燥,由此,得到27g的粉末状bis-af。
[0373]
《实施例2-3》
[0374]
首先,将bis-af(central glass公司制)200g置于具备搅拌马达、温度计及冷却冷凝器的硼硅酸玻璃制的3l烧瓶内,向其中注入正庚烷1800g及乙酸乙酯200g。
[0375]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0376]
在烧瓶的内温达到65℃后,花费1小时缓慢降温直至内温变为55℃,在该降温后,添加bis-af(central glass公司制)1g作为晶种,在该状态下搅拌1小时,使目标bis-af析出。
[0377]
进而,然后,冷却2小时直至内温变为35℃,通过减压过滤回收析出的结晶。在30℃下对回收的结晶进行减压干燥,由此,得到23g的粉末状bis-af。
[0378]
《实施例2-4》
[0379]
与上述的《实施例5》同样地进行操作,得到bis-af粉末。
[0380]
《比较例2-1》
[0381]
比较例2-1是通过依据专利文献2的实施例的方法来制造粉末的例(利用中和反应使bif-af析出)。
[0382]
将bis-af(central glass公司制)200g、氢氧化钠52g置于具备搅拌马达及温度计的硼硅酸玻璃制的3l烧瓶内。然后,一边注意放热,一边添加1800g水并进行搅拌,由此得到均匀的溶液(溶解有bif-af的盐)。
[0383]
接着,在冰浴中进行冷却直至内温变为10℃,然后,一边滴加35%盐酸水136g,一边进行中和,由此使bis-af析出。
[0384]
通过减压过滤回收析出的结晶,并在60℃下进行减压干燥。由此得到171g的粉末
状bis-af。
[0385]
《比较例2-2》
[0386]
比较例2-2是根据依据专利文献1的实施例的方法、条件来制造粉末的例(溶剂的主要成分为水)。
[0387]
将bis-af(central glass公司制)150g置于具备搅拌马达、温度计及冷却冷凝器的硼硅酸玻璃制的2l烧瓶内,向其中注入乙二醇300g及离子交换水700g。
[0388]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0389]
在烧瓶的内温达到65℃后,一边以10℃/小时的降温速度进行冷却直至烧瓶的内温变更为25℃,一边使bis-af析出。
[0390]
通过减压过滤回收析出的结晶,并在60℃下进行减压干燥。由此得到135g的粉末状bis-af。
[0391]
《比较例2-3》
[0392]
比较例2-3是根据依据专利文献(cn104529717a)的实施例的方法、条件来制造粉末的例(利用再沉淀使bis-af析出)。
[0393]
向具备搅拌马达、温度计的硼硅酸玻璃制的1l烧瓶内,注入离子交换水800g。在内温20~23℃下向其中滴加使bis-af(central glass公司制)100g溶解于100g甲醇得到的溶液,一边对烧瓶内进行搅拌,一边使bis-af再沉淀。
[0394]
通过减压过滤回收析出的结晶,并在60℃下进行减压干燥。由此得到89g的粉末状bis-af。
[0395]
《各种测定/评价》
[0396]
(粒径分布)
[0397]
利用岛津制作所制造的粒度分布计sald-2200,对使得到的粉末状bis-af分散于正癸烷溶剂中得到的样品的粒径分布进行测定。测定时,预先在载玻片上将bis-af与正癸烷溶剂混合,准备了糊剂状的bis-af。然后,向作为分散溶剂的正癸烷中一点点地添加糊剂状的bis-af,以吸光度的数值成为0.1l/(mol
·
cm)以下的方式调整添加量,对粒度分布进行测定。通过对得到的测定结果进行解析,计算出d
50
、d
90
、d
ave
及dm。
[0398]
将粒径的相关信息汇总示于表3中。
[0399]
[表3]
[0400]
表3
[0401][0402]
(操作性:块状化的评价)
[0403]
将bis-af粉末50g置于带盖的100ml聚乙烯制容器中,在室温下放置1个月。然后,
打开容器,取出全部的bis-af粉末,通过目视确认有无由bis-af的聚集所导致的块状化。
[0404]
将完全未确认到块状化的情况评价为
○
(良好),将确认到块状化的情况评价为
×
(差)。
[0405]
(操作性:流动性评价(漏斗试验))
[0406]
向漏斗口径150mm、喷嘴直径12mm、喷嘴长度22mm的漏斗投入全部的bis-af粉末60g,确认粉末能否自然排出。此时,将自然排出全部bis-af粉末的情况评价为〇(良好),将未自然排出全部bis-af粉末的情况评价为
×
(差)。
[0407]
(操作性:休止角的测定)
[0408]
使用漏斗口径150mm、喷嘴直径12mm、喷嘴长度22mm的漏斗,分别于漏斗上配置筛孔为0.5mm的筛板,在朝向下部距离漏斗喷嘴的前端4.5cm的位置上配置水平台。接着,取bis-af粉末50g至筛板上,使用刷子使bis-af粉末落向漏斗喷嘴的方向,使其堆积于水平台上。接着,对堆积的山状粉末拍摄照片,在照片上测定休止角。休止角越小,则表示粉末容易流畅地流动、即操作性优异。
[0409]
(堆积密度(松散堆积密度、振实堆积密度)的测定)
[0410]
使用筒井理化学器械制造的jis(japanese industrial standards,日本工业标准)堆积比重测定器,对堆积密度进行测定。堆积比重测定器分别具备漏斗口径150mm、喷嘴直径12mm、喷嘴长度22mm的漏斗;漏斗上的筛孔为0.5mm的筛板;及漏斗喷嘴的下部的圆筒状的30ml接收容器。取bis-af粉末50g至筛板上,使用刷子使其缓慢地落入接收容器,由此,使其自然填充。刮掉自接收容器溢出的bis-af粉末后,测定接收容器的重量,由此算出松散堆积密度。另外,使bis-af粉末落入时一边轻轻拍打接收容器的下部一边进行压缩填充,根据得到的重量计算出振实堆积密度。
[0411]
(溶剂溶解性/未溶解的评价)
[0412]
对于实施例2-1及比较例2-1~2-3的bis-af粉末,通过以下工序对溶剂溶解性进行评价。
[0413]
(i)在40%甲醇水溶液中的溶解速度的比较
[0414]
称取bis-af粉末10g,并将其置于具备搅拌子及温度计的硼硅酸玻璃制的100ml烧瓶内。在以均速搅拌粉末的bis-af的过程中,迅速注入40%甲醇水溶液40g。然后,维持均速的搅拌,在18~20℃下使bis-af粉末完全溶解。
[0415]
对上述工序中自刚添加甲醇水溶液后直至bis-af粉末完全溶解所花费的时间进行测定。
[0416]
(ii)在10%氢氧化钠水溶液中的溶解速度的比较
[0417]
量取10%氢氧化钠水溶液40g并将其置于具备搅拌子及温度计的硼硅酸玻璃制的100ml烧瓶内。向以均速搅拌的10%氢氧化钠水溶液中,花费30秒一点点地投入bis-af粉末10g。然后,维持均速的搅拌,测定直至在18~20℃下bis-af粉末完全溶解为止的累计时间。
[0418]
将各种测定/评价结果汇总示于表4中。
[0419]
[表4]
[0420][0421]
根据表3及表4,实施例2-1~2-4的粉末的块状化得到抑制。另外,实施例2-1~2-4的粉末的漏斗试验的结果良好。进而,实施例2-1~2-4的粉末的休止角小于比较例2-1及2-2的粉末。自这些结果可了解,d
50
为50~100μm,d
50
/d
ave
为1.1~1.5的化合物(a)的粉末具有良好的操作性。
[0422]
实施例2-1~2-4的粉末的堆积密度(松散堆积密度、振实堆积密度)大于比较例2-1~2-3的粉末。由此也可知,实施例2-1~2-3的粉末的操作性可谓良好。
[0423]
实施例2-1的粉末的溶剂溶解性良好。由此也可知,实施例2-1的粉末的操作性可谓良好。
[0424]
[实施例3-1~3-4及比较例3-1~3-3]
[0425]
以下的实施例3-1~3-4及比较例3-1~3-3特别是用于对第3实施方式详细进行说明的实施例及比较例。
[0426]
《实施例3-1》
[0427]
首先,将bis-af(central glass公司制)200g置于具备搅拌马达、温度计及冷却冷
凝器的硼硅酸玻璃制的3l烧瓶内,向其中注入正己烷1800g及乙酸乙酯200g。
[0428]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0429]
在烧瓶的内温达到65℃后,用1小时缓慢降温直至内温变为55℃,在该降温后,添加bis-af(central glass公司制)1g作为晶种,在该状态下搅拌1小时,使目标bis-af析出。
[0430]
进而,然后,冷却2小时直至内温变为35℃,通过减压过滤回收析出的结晶。在30℃下对回收的结晶进行减压干燥,由此,得到24g的粉末状bis-af。
[0431]
《实施例3-2》
[0432]
首先,将bis-af(central glass公司制)200g置于具备搅拌马达、温度计及冷却冷凝器的硼硅酸玻璃制的3l烧瓶内,向其中注入正己烷1800g及乙酸乙酯200g。
[0433]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0434]
在烧瓶的内温达到65℃后,用2小时缓慢降温直至内温变为55℃,在该降温后,添加bis-af(central glass公司制)1g作为晶种,在该状态下搅拌2小时,使目标bis-af析出。
[0435]
进而,然后,冷却4小时直至内温变为35℃,通过减压过滤回收析出的结晶。在30℃下对回收的结晶进行减压干燥,由此,得到27g的粉末状bis-af。
[0436]
《实施例3-3》
[0437]
首先,将bis-af(central glass公司制)200g置于具备搅拌马达、温度计及冷却冷凝器的硼硅酸玻璃制的3l烧瓶内,向其中注入正庚烷1800g及乙酸乙酯200g。
[0438]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0439]
在烧瓶的内温达到65℃后,用1小时缓慢降温直至内温变为55℃,在该降温后,添加bis-af(central glass公司制)1g作为晶种,在该状态下搅拌1小时,使目标bis-af析出。
[0440]
进而,然后,冷却2小时直至内温变为35℃,通过减压过滤回收析出的结晶。在30℃下对回收的结晶进行减压干燥,由此,得到23g的粉末状bis-af。
[0441]
《实施例3-4》
[0442]
与上述的《实施例5》同样地进行操作,得到bis-af粉末。
[0443]
《比较例3-1》
[0444]
比较例3-1是通过依据专利文献2的实施例的方法来制造粉末的例(利用中和反应使bif-af析出)。
[0445]
将bis-af(central glass公司制)200g、氢氧化钠52g置于具备搅拌马达及温度计的硼硅酸玻璃制的3l烧瓶内。然后,一边注意放热,一边添加1800g水并进行搅拌,由此得到均匀的溶液(溶解有bif-af的盐)。
[0446]
接着,在冰浴中进行冷却直至内温变为10℃,然后,一边滴加35%盐酸水136g,一边进行中和,由此使bis-af析出。
[0447]
通过减压过滤回收析出的结晶,并在60℃下进行减压干燥。由此得到171g的粉末状bis-af。
[0448]
《比较例3-2》
[0449]
比较例3-2是根据依据专利文献1的实施例的方法、条件来制造粉末的例(溶剂的
主要成分为水)。
[0450]
将bis-af(central glass公司制)150g置于具备搅拌马达、温度计及冷却冷凝器的硼硅酸玻璃制的2l烧瓶内,向其中注入乙二醇300g及离子交换水700g。
[0451]
然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为65℃,一边使bis-af溶解。
[0452]
在烧瓶的内温达到65℃后,一边以10℃/小时的降温速度进行冷却直至烧瓶的内温变更为25℃,一边使bis-af析出。
[0453]
通过减压过滤回收析出的结晶,并在60℃下进行减压干燥。由此得到135g的粉末状bis-af。
[0454]
《比较例3-3》
[0455]
比较例3-3是根据依据专利文献(cn104529717a)的实施例的方法、条件来制造粉末的例(利用再沉淀使bis-af析出)。
[0456]
向具备搅拌马达、温度计的硼硅酸玻璃制的1l烧瓶内,注入离子交换水800g。在内温20~23℃下向其中滴加使bis-af(central glass公司制)100g溶解于100g甲醇得到的溶液,一边对烧瓶内进行搅拌,一边使bis-af再沉淀。
[0457]
通过减压过滤回收析出的结晶,并在60℃下进行减压干燥。由此得到89g的粉末状bis-af。
[0458]
《各种测定/评价》
[0459]
(粒径分布)
[0460]
利用岛津制作所制造的粒度分布计sald-2200,对使得到的粉末状bis-af分散于正癸烷溶剂中得到的样品的粒径分布进行测定。测定时,预先在载玻片上将bis-af与正癸烷溶剂混合,准备了糊剂状的bis-af。然后,向作为分散溶剂的正癸烷中一点点地添加糊剂状的bis-af,以吸光度的数值成为0.1l/(mol
·
cm)以下的方式调整添加量,对粒度分布进行测定。通过对得到的测定结果进行解析,计算出d
50
、d
90
、d
ave
及dm。
[0461]
(休止角的测定)
[0462]
使用漏斗口径150mm、喷嘴直径12mm、喷嘴长度22mm的漏斗,分别于漏斗上配置筛孔为0.5mm的筛板,在朝向下部距离漏斗喷嘴的前端4.5cm的位置上配置水平台。接着,取bis-af粉末50g至筛板上,使用刷子使bis-af粉末落向漏斗喷嘴的方向,使其堆积于水平台上。接着,对堆积的山状粉末拍摄照片,在照片上测定休止角。
[0463]
(堆积密度(松散堆积密度、振实堆积密度)的测定)
[0464]
使用筒井理化学器械制造的jis堆积比重测定器,对堆积密度进行测定。堆积比重测定器分别具备漏斗口径150mm、喷嘴直径12mm、喷嘴长度22mm的漏斗;漏斗上的筛孔为0.5mm的筛板;及漏斗喷嘴的下部的圆筒状的30ml接收容器。取bis-af粉末50g至筛板上,使用刷子使其缓慢地落入接收容器,由此使其自然填充。刮掉自接收容器溢出的bis-af粉末后,测定接收容器的重量,由此算出松散堆积密度。另外,使bis-af粉末落入时一边轻轻拍打接收容器的下部一边进行压缩填充,根据得到的重量计算出振实堆积密度。
[0465]
(块状化的评价)
[0466]
将bis-af粉末50g置于带盖的100ml聚乙烯制容器中,在室温下放置1个月。然后,打开容器,取出全部的bis-af粉末,通过目视确认有无由bis-af的聚集所导致的块状化。
[0467]
将完全未确认到块状化的情况评价为
○
(良好),将确认到块状化的情况评价为
×
(差)。
[0468]
(流动性评价:漏斗试验)
[0469]
向漏斗口径150mm、喷嘴直径12mm、喷嘴长度22mm的漏斗投入全部的bis-af粉末60g,确认粉末能否自然排出。此时,将自然排出全部bis-af粉末的情况评价为〇(良好),将未自然排出全部bis-af粉末的情况评价为
×
(差)。
[0470]
(溶剂溶解性/未溶解的评价)
[0471]
对于实施例3-1及比较例3-1~3-4的bis-af粉末,通过以下工序对溶剂溶解性进行评价。
[0472]
(i)在40%甲醇水溶液中的溶解速度的比较
[0473]
称取bis-af粉末10g,并将其置于具备搅拌子及温度计的硼硅酸玻璃制的100ml烧瓶内。在以均速搅拌粉末的bis-af的过程中,迅速注入40%甲醇水溶液40g。然后,维持均速的搅拌,在18~20℃下使bis-af粉末完全溶解。
[0474]
对上述工序中自刚添加甲醇水溶液后直至bis-af粉末完全溶解所花费的时间进行测定。
[0475]
(ii)在10%氢氧化钠水溶液中的溶解速度的比较
[0476]
量取10%氢氧化钠水溶液40g并将其置于具备搅拌子及温度计的硼硅酸玻璃制的100ml烧瓶内。向以均速搅拌的10%氢氧化钠水溶液中,花费30秒一点点地投入bis-af粉末10g。然后,维持均速的搅拌,测定直至在18~20℃下bis-af粉末完全溶解为止的累计时间。
[0477]
将上述各种信息汇总示于表5及表6中。
[0478]
[表5]
[0479][0480]
[表6]
[0481]
表6
[0482][0483]
根据表5及表6,实施例3-1~3-4的粉末的块状化及漏斗试验的相关评价结果良好。另外,实施例3-1的粉末的溶剂溶解性良好。即,d
50
为50~100μm、休止角为35~49
°
的化合物(a)的粉末在工业上的操作性优异。
[0484]
[实施例4-1~4-5及比较例4-1~4-3]
[0485]
以下的实施例4-1~4-5及比较例4-1~4-3特别是用于对第4实施方式详细进行说明的实施例及比较例。
[0486]
《实施例4-1》
[0487]
自试剂瓶采集东京化成工业株式会社制造的bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水202.5g及甲醇22.5g,一边搅拌一边将容器内部的温度升温至65℃。
[0488]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下维持温度,并搅拌1.5小时。
[0489]
经过1.5小时后,暂时停止搅拌并静置,对容器内进行目视观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0490]
此时,使用对-双(三氟甲基)苯作为内标,通过19f-nmr(nuclear magnetic resonance,核磁共振)对水层侧的bis-af浓度进行测定。浓度为2质量%以下,尽管为65℃左右的高温,大部分bis-af也未“溶解”于水/甲醇中。即,在原料的bis-af粉末会完全“熔解”的温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0491]
接着,一边将内温维持在60~65℃的温度一边搅拌1小时,结果结晶析出。然后,将内温以15℃/1小时的降温速度冷却至室温。需要说明的是,此时的室温约为25℃。
[0492]
冷却后,使用具备滤纸的吸滤器,通过减压过滤分离并回收析出的bis-af。回收后用75ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在80℃、减压(1kpa以下)下干燥6小时。
[0493]
干燥后得到的bis-af粉末为22.9g,收率为92%。
[0494]
《实施例4-2》
[0495]
将bis-af粉末的量变更为100.0g(298mmol),将纯水的量变更为810.0g,将甲醇的
量变更为90.0g,除此以外,与实施例4-1同样地得到bis-af粉末。
[0496]
干燥后得到的bis-af粉末为94.6g,收率为95%。
[0497]
需要说明的是,与实施例1同样,经过1.5小时后,暂时停止搅拌并静置,对容器内进行目视观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0498]
此时,使用对-双(三氟甲基)苯作为内标,通过19f-nmr对水层侧的bis-af浓度进行测定。浓度为2质量%以下,尽管为65℃左右的高温,大部分bis-af也未“溶解”于水/甲醇中。即,在原料的bis-af粉末会完全“熔解”的温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0499]
《实施例4-3》
[0500]
自与实施例4-1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水225.0g。此外,一边搅拌一边将容器内部的温度升温至95℃。
[0501]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下将温度维持在95℃并搅拌1小时。
[0502]
经过1小时后,暂时停止搅拌并静置,对容器内进行目视观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0503]
此时,使用对-双(三氟甲基)苯作为内标,通过19f-nmr对水层侧的bis-af浓度进行测定。浓度为2质量%左右,尽管为95℃左右的高温,大部分bis-af也未“溶解”于水中。即,在原料的bis-af粉末的熔解温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0504]
接着,一边将内温维持在92~94℃,一边搅拌1小时,结果结晶析出。然后,将内温以15℃/1小时的降温速度冷却至室温。此时的室温约为25℃。
[0505]
冷却后,针对析出的bis-af,使用具备滤纸的吸滤器,通过减压过滤进行分离并回收。回收后用75ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在75℃、减压(1kpa以下)下干燥8小时。
[0506]
干燥后得到的bis-af粉末为24.1g,收率为96%。
[0507]
《实施例4-4》
[0508]
自与实施例4-1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水112.5g及甲醇12.5g,一边搅拌一边将容器内部的温度升温至65℃。
[0509]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下维持温度,并搅拌1.5小时。
[0510]
经过1.5小时后,暂时停止搅拌并静置,对容器内进行目视观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0511]
此时,使用对-双(三氟甲基)苯作为内标,通过19f-nmr对水层侧的bis-af浓度进行测定。浓度为2质量%以下,尽管为65℃左右的高温,大部分bis-af也未“溶解”于水/甲醇
中。即,在原料的bis-af粉末会完全“熔解”的温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0512]
接着,一边将内温维持在60~65℃的温度一边搅拌1小时,结果结晶析出。然后,将内温以15℃/1小时的降温速度冷却至室温。需要说明的是,此时的室温约为25℃。
[0513]
冷却后,使用具备滤纸的吸滤器,通过减压过滤分离并回收析出的bis-af。回收后用75ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在80℃、减压(1kpa以下)下干燥6小时。
[0514]
干燥后得到的bis-af粉末为23.5g,收率为94%。
[0515]
《实施例4-5》
[0516]
自与实施例4-1相同的试剂瓶采集bis-af粉末25.0g(74.4mmol),并将其置于具备搅拌机及冷却冷凝器的容积为500ml的硼硅酸玻璃制容器中。接着,向容器内添加纯水100.0g及甲醇25.0g,一边搅拌一边将容器内部的温度升温至45℃。
[0517]
在升温期间,bis-af粉末开始熔解,容器内变为悬浮状态。在该状态下维持温度,并搅拌1.5小时。
[0518]
经过1.5小时后,暂时停止搅拌并静置,对容器内进行目视观察。观察的结果为,确认到在容器内两相即水层与bis-af的熔解物分离(不完全地混合)地存在。另外,未确认到未熔解的bis-af。
[0519]
此时,使用对-双(三氟甲基)苯作为内标,通过19f-nmr对水层侧的bis-af浓度进行测定。浓度为2质量%以下,尽管为45℃左右的温度,大部分bis-af也未“溶解”于水/甲醇中。即,在原料的bis-af粉末会完全“熔解”的温度下,水系分散介质的bis-af的溶解度大幅低于10[g/100g]。
[0520]
接着,一边将内温维持在35~40℃的温度一边搅拌1小时,结果结晶析出。然后,将内温以10℃/1小时的降温速度冷却至室温。需要说明的是,此时的室温约为25℃。
[0521]
冷却后,使用具备滤纸的吸滤器,通过减压过滤分离并回收析出的bis-af。回收后用75ml的纯水进行挂洗。使用真空干燥器将清洗过的bis-af在80℃、减压(1kpa以下)下干燥6小时。
[0522]
干燥后得到的bis-af粉末为24.1g,收率为96%。
[0523]
《比较例4-1》
[0524]
自试剂瓶采集bis-af(东京化成工业株式会社制)。
[0525]
《比较例4-2》
[0526]
向具备搅拌机的容积为500l的硼硅酸玻璃制容器中注入纯水200g,在室温(约20℃)下开始搅拌。利用滴加漏斗,向其中缓慢滴加溶解于甲醇50g得到的bis-af(东京化成工业株式会社制)25.0g,进行bis-af的再沉淀。
[0527]
bis-af析出后,使用具备滤纸的吸滤器,通过减压过滤回收bis-af。接着,使用真空干燥器,在80℃、减压下干燥8小时。干燥后得到的bis-af粉末为22.1g,收率为88%。
[0528]
《比较例4-3》
[0529]
将bis-af(东京化成工业株式会社制)150g置于具备搅拌机及冷却冷凝器的容积为2l的硼硅酸玻璃制容器中,向其中注入乙二醇300g及离子交换水700g。然后,一边使用油浴进行加热搅拌直至烧瓶的内温变更为85℃,一边使bis-af溶解。在该状态下维持温度,并
搅拌1小时,经过1小时后,暂时停止搅拌并静置,对容器内进行目视观察,其结果确认溶液均匀。
[0530]
接着,一边搅拌,一边以10℃/小时的降温速度进行冷却直至烧瓶的内温变更为25℃,其结果,确认降温过程中结晶析出。通过减压过滤回收析出的结晶,并在60℃下进行减压干燥。干燥后得到的bis-af粉末为137g,收率为91%。
[0531]
《测定》
[0532]
(x射线衍射(xrd)光谱)
[0533]
粉末的x射线衍射(xrd)光谱基于上述测定方法及测定条件。
[0534]
将x射线衍射光谱的相关信息汇总示于表7中。
[0535]
[表7]
[0536][0537]
《评价》
[0538]
(溶解速度)
[0539]
(i)在40%甲醇水溶液(40%meoh水)中的溶解速度的比较
[0540]
称取bis-af粉末10g,并将其置于具备搅拌子及温度计的硼硅酸玻璃制的100ml烧瓶内。在以均速(200rpm)搅拌粉末的bis-af的过程中,迅速注入40%甲醇水溶液40g。然后,维持在均速(200rpm)下的搅拌,在18~20℃下使bis-af粉末完全溶解。
[0541]
对上述工序中自刚添加甲醇水溶液后直至bis-af粉末完全溶解所花费的时间进行测定。bis-af粉末完全熔解是通过目视观察进行确认。
[0542]
(ii)在10%氢氧化钠水溶液(10%naoh水)中的溶解速度的比较
[0543]
量取10%氢氧化钠水溶液40g并将其置于具备搅拌子及温度计的硼硅酸玻璃制的100ml烧瓶内。向以均速(200rpm)搅拌的10%氢氧化钠水溶液中,花费30秒一点点地投入
bis-af粉末10g。然后,维持在均速(200rpm)下的搅拌,对刚开始添加bis-af粉末后至在18~20℃下bis-af粉末完全溶解的累计时间进行测定。bis-af粉末完全熔解是通过目视观察进行确认的。
[0544]
(干燥试验)
[0545]
使bis-af粉末分散于水中的分散液通过具备滤纸的过滤器而进行过滤,得到被水弄湿的bis-af粉末。将得到的bis-af粉末置于yamato scientific株式会社制造的真空干燥机中,在0.5kpa、60℃下进行干燥,对粉末的含水率进行测定。
[0546]
放入bis-af粉末10g,通过容量滴定式卡尔费休水份滴定仪(mkv-710b,京都电子工业株式会社制)对于0.5kpa、60℃下开始干燥的时点(0小时)、0.5小时后、1小时后的粉末的含水率进行测定。
[0547]
将评价结果汇总示于下表。
[0548]
[表8]
[0549][0550]
n/a表示“无数据”。
[0551]
如表8所示,实施例4-1、4-4及4-5的bis-af的粉末的溶解速度良好。另外,自含水率的评价结果可知,对实施例4-1、4-4及4-5的bis-af的粉末进行干燥时,干燥时间较短即可。即,实施例4-1等的bis-af的粉末在工业上的操作性优异。
[0552]
另外,对实施例4-1,如下进行“吸湿试验”。
[0553]
(吸湿试验)
[0554]
在培养皿中称取bis-af粉末10g,通过容量式卡尔费休水份滴定仪(mkv-710b,京都电子工业株式会社制)对bis-af粉末的含水率进行测定。
[0555]
另外,在培养皿中称取bis-af粉末10g,将其静置于温度30℃、湿度98%的恒温恒湿槽内,经过24小时后,通过容量式卡尔费休水份滴定仪(mkv-710b,京都电子工业株式会社制)对bis-af粉末的含水率进行测定。
[0556]
将评价结果表示于下表。
[0557]
[表9]
[0558][0559]
如表9所示,可知实施例4-1的bis-af的粉末其含水率24小时后也不变,不易吸湿。
即,实施例4-1的bis-af粉末在工业上的操作性优异。
[0560]
该申请主张以2019年9月30日申请的日本技术特愿2019-178923号、2019年9月30日申请的日本技术特愿2019-178924号、2019年12月27日申请的日本技术特愿2019-238016号及2020年2月28日申请的日本技术特愿2020-033388号为基础的优先权,将其公开内容全部引入至此。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。