1.本发明涉及化学合成领域,具体为一类大麻酚衍生物化合物及其制备方法。
背景技术:
2.大麻酚是一种麻醉药,分子式c
21h26
o2,它存在于大麻叶中,有止咳、镇痉、止痛、镇静、安眠等活性,为一种嗜好品,吸食后产生精神愉快感,能致习惯性,应用时应小心。大麻叶中含有多种大麻酚类衍生物,已分离到15种以上,较重要的有:大麻酚、大麻二酚、四氢大麻酚、大麻酚酸、大麻二酚酸、四氢大麻酚酸。
3.大麻酚(cannabinol,cbn)可通过非酶促反应经大麻酚酸脱除羧基转化而来。研究发现cbn类似物可以选择性地与cb2受体作用,从而起到抗炎、抗疼痛以及抑制癌细胞扩散等药理作用。但是此领域的研究还不成熟,亟待进一步的挖掘和探索,所以研究大麻酚的衍生物及其合成方法具有十分重大的意义。
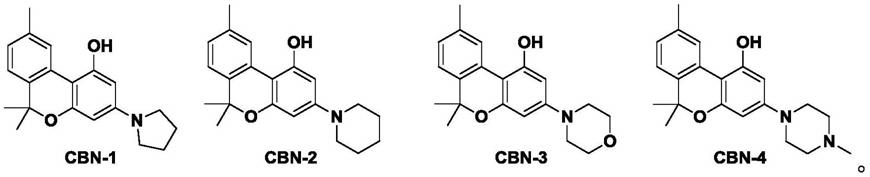
4.我们设计和合成了一类大麻酚的衍生物(cbn-1,cbn-2,cbn-3和cbn-4)。四种化合物通过在大麻酚的分子结构中分别引入多元环结构,从而有利于提高药物的药效。
技术实现要素:
5.本发明目的在于提供一类大麻酚衍生物化合物及其制备方法,能够对大麻酚的结构进行修饰;由于引入不同的药效基团,我们认为这使得该类衍生物相比于大麻酚具有更高的药效。
6.为达成上述目的,本发明提出如下技术方案:一类大麻酚衍生物化合物,包括cbn-1、cbn-2、cbn-3和cbn-4,结构式分别如下:
[0007][0008]
一类大麻酚衍生物化合物的制备方法,所述cbn-1、cbn-2、cbn-3和cbn-4的合成方法反应式如下:
[0009]
[0010]
进一步的,在本发明中,所述cbn-1的合成方法具体步骤如下:
[0011]
向厚壁耐压瓶中加入(e)-1-(3-甲氧基-4-(3-甲基丁烷-1,3-二烯-1-基)苯基)吡咯烷和丙炔酸甲酯,然后加入乙醇溶液中,反应加热至80℃后继续反应8小时,待反应液冷却至室温,减压浓缩,加入乙酸乙酯和水,分液得到有机相,然后水相用乙酸乙酯萃取两次,合并有机相,有机相经无水硫酸钠干燥后,硅胶柱层析纯化,所得白色固体即为cbn-1-compound-1;
[0012]
将cbn-1-compound-1溶解在乙酸酐溶液中,然后加入碘化氢溶液,反应加热至140℃后反应1小时,待反应液冷却至70℃后倒水冰水中,向上述溶液中加入乙酸乙酯,分离有机相,然后水相用乙酸乙酯萃取两次,合并有机相,有机相经无水硫酸钠干燥后,减压浓缩,除去残留溶剂,所得粗品经硅胶柱层析纯化,所得灰色固体即为cbn-1-compound-2;
[0013]
将cbn-1-compound-2溶解在四氢呋喃溶液中,然后加入甲基溴化镁,反应加热至66℃后反应1小时,待反应液冷却至室温后倒水冰的氯化铵饱和溶液中,向上述溶液中加入甲苯,分离有机相,然后水相用甲苯萃取两次,合并有机相,有机相经无水硫酸钠干燥后,加入对甲苯磺酸,溶液在110℃回流3小时后,减压浓缩出去甲苯,向所得粗品中加入甲苯和乙醇的比例为4:1的混合溶剂回流1小时后,待溶液冷却至室温置于4度冰箱8小时,抽滤所得溶液,所得灰色固体即为cbn-1。
[0014]
有益效果,本技术的技术方案具备如下技术效果:
[0015]
本发明提供一类大麻酚衍生物化合物及其制备方法,其在大麻酚的结构中分别引入氮杂环丁烷、哌啶环、吗啉环和4-甲基哌嗪环,从而提高其药效。
[0016]
应当理解,前述构思以及在下面更加详细地描述的额外构思的所有组合只要在这样的构思不相互矛盾的情况下都可以被视为本公开的发明主题的一部分。
[0017]
结合附图从下面的描述中可以更加全面地理解本发明教导的前述和其他方面、实施例和特征。本发明的其他附加方面例如示例性实施方式的特征和/或有益效果将在下面的描述中显见,或通过根据本发明教导的具体实施方式的实践中得知。
附图说明
[0018]
附图不意在按比例绘制。在附图中,在各个图中示出的每个相同或近似相同的组成部分可以用相同的标号表示。为了清晰起见,在每个图中,并非每个组成部分均被标记。现在,将通过例子并参考附图来描述本发明的各个方面的实施例,其中:
[0019]
图1为本发明的大麻酚衍生物化合物及其制备方法。
具体实施方式
[0020]
为了更了解本发明的技术内容,特举具体实施例并配合所附图式说明如下。在本公开中参照附图来描述本发明的各方面,附图中示出了许多说明的实施例。本公开的实施例不必定义在包括本发明的所有方面。应当理解,上面介绍的多种构思和实施例,以及下面更加详细地描述的那些构思和实施方式可以以很多方式中任意一种来实施,这是因为本发明所公开的构思和实施例并不限于任何实施方式。另外,本发明公开的一些方面可以单独使用,或者与本发明公开的其他方面的任何适当组合来使用。
[0021]
实施例1:一类大麻酚衍生物(cbn-1)的结构及制备方法:
[0022][0023]
cbn-1-compound-1:向厚壁耐压瓶中加入1g(e)-1-(3-甲氧基-4-(3-甲基丁烷-1,3-二烯-1-基)苯基)吡咯烷和0.7g丙炔酸甲酯,然后加入50ml乙醇溶液中。反应加热至80℃后继续反应8小时。待反应液冷却至室温,减压浓缩,加入50ml乙酸乙酯和50ml水。分液得到有机相,然后水相用50ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,硅胶柱层析纯化,所得白色固体即为cbn-1-compound-1,产量0.72g,产率为42.4%,纯度为99.2%。1h nmr(400mhz,cdcl3)δ2.09(t,j=5.2hz,4h),2.46(s,3h),3.43(t,j=7.9hz,4h),3.81(s,6h),3.95(s,3h),6.23(s,2h),7.39(d,j=3.2hz,1h),7.87(s,1h),8.42(d,j=1.2hz,1h)。
[0024]
cbn-1-compound-2:将5g cbn-1-compound-1溶解在200ml乙酸酐溶液中,然后加入10ml碘化氢溶液,反应加热至140℃后反应1小时,待反应液冷却至70℃后倒水冰水中。向上述溶液中加入200ml乙酸乙酯,分离有机相。然后水相用100ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,减压浓缩,除去残留溶剂。所得粗品经硅胶柱层析纯化。所得灰色固体即为cbn-1-compound-2,产量为4.18g,产率为83.6%,纯度为99.1%。1h nmr(400mhz,cdcl3)δ2.05(t,j=5.1hz,4h),2.47(s,3h),3.41(t,j=7.9hz,4h),6.23(s,1h),6.39(s,1h),6.97(d,j=3.2hz,1h),7.37(d,j=6.2hz,1h),7.87(s,1h),9.42(s,1h)。
[0025]
cbn-1:1g cbn-1-compound-2溶解在50ml四氢呋喃溶液中,然后加入5ml甲基溴化镁,反应加热至66℃后反应1小时,待反应液冷却至室温后倒水冰的氯化铵饱和溶液中。向上述溶液中加入100ml甲苯,分离有机相。然后水相用50ml甲苯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,加入10ml对甲苯磺酸。溶液在110℃回流3小时后,减压浓缩出去甲苯。向所得粗品中加入甲苯和乙醇的比例为4:1的混合溶剂回流1小时后,待溶液冷却至室温置于4度冰箱8小时。抽滤所得溶液,所得灰色固体即为cbn-1,产量为0.78g,产率为62.0%,纯度为98.3%。1h nmr(400mhz,cdcl3)δ1.58(s,6h),2.19(t,j=5.2hz,4h),2.46(s,3h),3.43(t,j=7.9hz,4h),5.23(s,1h),6.09(s,1h),6.12(s,1h),7.27(d,j=2.2hz,1h),7.42(d,j=1.2hz,1h),7.82(s,1h),9.61(s,1h).
[0026]
实施例2:一类大麻酚衍生物(cbn-2)的结构及制备方法:
[0027][0028]
cbn-2-compound-1:向厚壁耐压瓶中加入1g(e)-1-(3-甲氧基-4-(3-甲基丁烷-1,3-二烯-1-基)苯基)哌啶和0.71g丙炔酸甲酯,然后加入50ml乙醇溶液中。反应加热至80℃后继续反应8小时。待反应液冷却至室温,减压浓缩,加入50ml乙酸乙酯和50ml水。分液得到有机相,然后水相用50ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,硅胶柱层析纯化,所得白色固体即为cbn-2-compound-1,产量0.69g,产率为40.4%,纯度为99.2%。1h nmr(400mhz,cdcl3)δ2.09(t,j=5.3hz,4h),2.48(s,3h),3.53(m,j=6.9hz,1h),3.43(m,j=7.9hz,4h),3.86(s,6h),3.96(s,3h),6.25(s,2h),7.29(d,j=3.2hz,1h),7.87(s,1h),8.48(d,j=5.4hz,1h)。
[0029]
cbn-2-compound-2:将5g cbn-1-compound-1溶解在200ml乙酸酐溶液中,然后加入10ml碘化氢溶液,反应加热至140℃后反应1小时,待反应液冷却至70℃后倒水冰水中。向上述溶液中加入200ml乙酸乙酯,分离有机相。然后水相用100ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,减压浓缩,除去残留溶剂。所得粗品经硅胶柱层析纯化。所得灰色固体即为cbn-2-compound-2,产量为2.29g,产率为45.8%,纯度为99.1%。1h nmr(400mhz,cdcl3)δ2.05(t,j=5.1hz,4h),2.47(s,3h),2.61(m,j=7.9hz,1h),3.41(t,j=7.9hz,4h),6.26(s,1h),6.37(s,1h),6.96(d,j=3.2hz,1h),7.38(d,j=6.8hz,1h),7.88(s,1h),9.45(s,1h)。
[0030]
cbn-2:1g cbn-2-compound-2溶解在50ml四氢呋喃溶液中,然后加入5ml甲基溴化镁,反应加热至66℃后反应1小时,待反应液冷却至室温后倒水冰的氯化铵饱和溶液中。向上述溶液中加入100ml甲苯,分离有机相。然后水相用50ml甲苯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,加入10ml对甲苯磺酸。溶液在110℃回流3小时后,减压浓缩出去甲苯。向所得粗品中加入甲苯和乙醇的比例为4:1的混合溶剂回流1小时后,待溶液冷却至室温置于4度冰箱8小时。抽滤所得溶液,所得灰色固体即为cbn-2,产量为0.68g,产率为45.3%,纯度为99.3%。1h nmr(400mhz,cdcl3)δ1.52(s,6h),2.09(m,1h),2.29(m,j=5.2hz,4h),2.13(s,3h),3.12(t,j=7.9hz,4h),5.25(s,1h),6.37(s,1h),6.45(s,1h),7.27(d,j=2.3hz,1h),7.42(d,j=1.5hz,1h),7.85(s,1h),9.62(s,1h)。
[0031]
实施例3:一类大麻酚衍生物(cbn-3)的结构及制备方法:
[0032][0033]
cbn-3-compound-1:向厚壁耐压瓶中加入1g(e)-1-(3-甲氧基-4-(3-甲基丁烷-1,3-二烯-1-基)苯基)吗啉和0.72g丙炔酸甲酯,然后加入50ml乙醇溶液中。反应加热至80℃后继续反应8小时。待反应液冷却至室温,减压浓缩,加入50ml乙酸乙酯和50ml水。分液得到有机相,然后水相用50ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,硅胶柱层析纯化,所得白色固体即为cbn-3-compound-1,产量0.83g,产率为48.3%,纯度为99.2%。1h nmr(400mhz,cdcl3)δ2.09(t,j=5.3hz,4h),2.47(s,3h),3.43(t,j=5.9hz,4h),3.82(s,6h),3.96(s,3h),6.13(s,2h),7.49(d,j=6.4hz,1h),7.88(s,1h),8.52(d,j=8.2hz,1h)。
[0034]
cbn-3-compound-2:将5g cbn-3-compound-1溶解在200ml乙酸酐溶液中,然后加入10ml碘化氢溶液,反应加热至140℃后反应1小时,待反应液冷却至70℃后倒水冰水中。向上述溶液中加入200ml乙酸乙酯,分离有机相。然后水相用100ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,减压浓缩,除去残留溶剂。所得粗品经硅胶柱层析纯化。所得灰色固体即为cbn-3-compound-2,产量为4.41g,产率为88.2%,纯度为99.1%。1h nmr(400mhz,cdcl3)δ2.15(t,j=5.1hz,4h),2.27(s,3h),3.51(t,j=1.2hz,4h),6.13(s,1h),6.29(s,1h),6.87(d,j=2.2hz,1h),7.35(d,j=1.2hz,1h),7.88(s,1h),9.43(s,1h).
[0035]
cbn-3:1g cbn-3-compound-2溶解在50ml四氢呋喃溶液中,然后加入5ml甲基溴化镁,反应加热至66℃后反应1小时,待反应液冷却至室温后倒水冰的氯化铵饱和溶液中。向上述溶液中加入100ml甲苯,分离有机相。然后水相用50ml甲苯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,加入10ml对甲苯磺酸。溶液在110℃回流3小时后,减压浓缩出去甲苯。向所得粗品中加入甲苯和乙醇的比例为4:1的混合溶剂回流1小时后,待溶液冷却至室温置于4度冰箱8小时。抽滤所得溶液,所得灰色固体即为cbn-3,产量为0.74g,产率为49.3%,纯度为98.9%。1h nmr(400mhz,cdcl3)δ1.53(s,6h),2.30(t,j=5.2hz,4h),2.23(s,3h),3.22(t,j=7.8hz,4h),5.15(s,1h),6.27(s,1h),6.35(s,1h),7.49(d,j=2.3hz,1h),7.48(d,j=1.5hz,1h),7.87(s,1h),9.42(s,1h)。
[0036]
实施例4:一类大麻酚衍生物(cbn-4)的结构及制备方法:
[0037][0038]
cbn-4-compound-1:向厚壁耐压瓶中加入1g(e)-1-(3-甲氧基-4-(3-甲基丁烷-1,3-二烯-1-基)苯基)-4-甲基哌嗪和0.72g丙炔酸甲酯,然后加入50ml乙醇溶液中。反应加热至80℃后继续反应8小时。待反应液冷却至室温,减压浓缩,加入50ml乙酸乙酯和50ml水。分液得到有机相,然后水相用50ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,硅胶柱层析纯化,所得白色固体即为cbn-4-compound-1,产量0.83g,产率为48.3%,纯度为99.2%。1h nmr(400mhz,cdcl3)δ2.19(t,j=5.3hz,4h),2.27(s,3h),3.28(s,3h),3.53(t,j=8.9hz,4h),3.83(s,6h),3.98(s,3h),6.33(s,2h),7.59(d,j=8.4hz,1h),7.78(s,1h),8.92(d,j=8.5hz,1h)。
[0039]
cbn-4-compound-2:将5g cbn-4-compound-1溶解在200ml乙酸酐溶液中,然后加入10ml碘化氢溶液,反应加热至140℃后反应1小时,待反应液冷却至70℃后倒水冰水中。向上述溶液中加入200ml乙酸乙酯,分离有机相。然后水相用100ml乙酸乙酯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,减压浓缩,除去残留溶剂。所得粗品经硅胶柱层析纯化。所得灰色固体即为cbn-1-compound-2,产量为4.41g,产率为88.2%,纯度为99.1%。1h nmr(400mhz,cdcl3)δ2.15(t,j=5.1hz,4h),2.17(s,3h),2.57(s,3h),3.61(t,j=4.2hz,4h),6.23(s,1h),6.39(s,1h),6.89(d,j=2.9hz,1h),7.45(d,j=4.2hz,1h),7.86(s,1h),9.23(s,1h).
[0040]
cbn-4:1g cbn-4-compound-2溶解在50ml四氢呋喃溶液中,然后加入5ml甲基溴化镁,反应加热至66℃后反应1小时,待反应液冷却至室温后倒水冰的氯化铵饱和溶液中。向上述溶液中加入100ml甲苯,分离有机相。然后水相用50ml甲苯萃取两次,合并有机相。有机相经无水硫酸钠干燥后,加入10ml对甲苯磺酸。溶液在110℃回流3小时后,减压浓缩出去甲苯。向所得粗品中加入甲苯和乙醇的比例为4:1的混合溶剂回流1小时后,待溶液冷却至室温置于4度冰箱8小时。抽滤所得溶液,所得灰色固体即为cbn-3,产量为0.74g,产率为49.3%,纯度为98.9%。1h nmr(400mhz,cdcl3)δ1.53(s,6h),2.03(s,3h)2.31(t,j=5.1hz,4h),2.13(s,3h),3.22(t,j=7.3hz,4h),5.65(s,1h),6.97(s,1h),6.25(s,1h),7.29(d,j=2.3hz,1h),7.58(d,j=1.4hz,1h),7.89(s,1h),9.42(s,1h)。
[0041]
虽然本发明已以较佳实施例揭露如上,然其并非用以限定本发明。本发明所属技术领域中具有通常知识者,在不脱离本发明的精神和范围内,当可作各种的更动与润饰。因此,本发明的保护范围当视权利要求书所界定者为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。