1.本发明涉及卡利拉嗪及其中间体的制备方法。
背景技术:
2.2015年,美国fda批准卡利拉嗪(cariprazine;商品名:vraylar)胶囊用于治疗精神分裂症和双相情感障碍成人患者。卡利拉嗪(criprazine)为多巴胺d3/d2受体部分激动剂,是一种试验性非典型抗精神病用药,用于精神分裂症患者和双相ⅰ型障碍相关的躁狂或混合发作患者。
[0003][0004]
专利wo2005012266最早报道了卡利拉嗪的合成,首先将1-(2,3-二氯苯基)哌嗪和反式2-{1-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙醛反应还原胺化得到反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己基}-氨基甲酸叔丁酯。再经脱叔丁氧羰基保护,与二甲基-氨基甲酰氯反应得卡利拉嗪。原料反式2-{1-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙醛的制备由journal of medicinal chemistry,2000,vol.43.9.1878-1885描述的还原方法获得,该还原的主要缺点是必须在-78℃无水甲苯中将dibal-h试剂加入反应混合物,操作危险,对仪器设备要求高,工业化生产困难。合成路线如下:
[0005][0006]
专利wo2010070369描述了用2-{反式-4-[(叔丁氧基羰基)氨基]环己基}乙基甲烷磺酸酯进行n-烷基化的路线。2-{反式-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙酸乙酯经还原反应并随后与甲烷磺酰氯反应得到2-{反式-4-[(叔丁氧基羰基)氨基]环己基}乙基甲烷磺酸酯,与1-(2,3-二氯苯基)哌嗪发生n-烷基化反应得n-叔丁氧基羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺,再经脱叔丁氧羰基保护,得到关键中间体4-{2-[-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺二盐酸盐一水合物。该过程的缺点是甲磺酸酯衍生物余留在反应混合物中,其被视为潜在的遗传毒性污染,并且污染程度保持在ppm水平,而且甲烷磺酰氯在生产使用过程中毒性较大,不利于工业生产和劳动保护。合成路线如
下:
[0007][0008]
专利wo2018007986描述了卡利拉嗪的新制备方法,根据该过程中以反式2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐为起始原料,经水解、酸化,得到反式2-[1-(4-氨基]-环己基)-乙酸盐酸盐,然后与二甲基-氨基甲酰氯反应得到反式-4-{[(二甲基氨基羰基]氨基}环己基)乙酸,然后将反式-4-{[(二甲基氨基羰基]氨基}环己基)乙酸与1-(2,3-二氯苯基)-哌嗪经由酰氯或者n,n-羰基二咪唑缩合得1-二甲基-3-[反式-4-(2-氧代-2-(4-(2,3-二氯苯基)哌嗪-1-基-乙基)环己基]脲,然后经硼氢化钠和三氟化硼-乙醚合物或硼氢化钠和无水三氯化铝将酰胺键还原得到卡利拉嗪,该路线整体收率很低。合成路线如下:
[0009][0010]
目前,急需开发一种操作简单安全、收率高、适合于工业化生产的卡利拉嗪及其中间体的制备方法。
技术实现要素:
[0011]
本发明为了克服现有技术中卡利拉嗪的制备方法反应步骤繁琐、操作危险、设备腐蚀性强、收率不高、不适合于工业化生产等缺陷,而提供了一种卡利拉嗪及其中间体的制备方法。本发明的制备方法,原料易得、反应步骤短、操作简单安全、环境友好、后处理简单、收率高、制得的产品纯度高、适合于工业化生产。
[0012]
本发明提供了一种卡利拉嗪中间体5,其结构如下所示:
[0013][0014]
其中,pg为氨基保护基或氢。所述的氨基保护基优选叔丁氧羰基或乙酰基等。
[0015]
本发明还提供了所述的卡利拉嗪中间体5的制备方法,其包括以下步骤:
[0016][0017]
a)溶剂中,碱存在下,将2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1与氨基保护试剂进行缩合反应,得到卡利拉嗪中间体2;
[0018]
b)在溶剂中,将步骤a)得到的卡利拉嗪中间体2,在碱性条件水解,然后酸化,得到卡利拉嗪中间体3;
[0019]
c)有机溶剂中,催化剂和碱存在下,将步骤b)得到的卡利拉嗪中间体3与1-(2,3-二氯苯基)-哌嗪4进行缩合反应,得到卡利拉嗪中间体5即可。
[0020]
步骤a)中,所述的溶剂优选水和/或有机溶剂。所述的有机溶剂优选卤代烃类溶剂、醇类溶剂、酮类溶剂、醚类溶剂、酯类溶剂、腈类溶剂、芳烃类溶剂和酰胺类溶剂的一种或多种。所述的卤代烃类溶剂优选氯代烃类溶剂;所述的氯代烃类溶剂优选二氯甲烷、二氯乙烷和三氯甲烷中的一种或多种。所述的醇类溶剂优选甲醇、乙醇和叔丁醇中的一种或多种。所述的酮类溶剂优选丙酮。所述的醚类溶剂优选四氢呋喃和/或1,4-二氧六环。所述的酯类溶剂优选乙酸乙酯。所述的腈类溶剂优选乙腈。所述的芳烃类溶剂优选甲苯。所述的酰胺类溶剂优选n,n-二甲基甲酰胺。
[0021]
步骤a)中,所述的溶剂与所述的2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1的体积质量比值优选1ml/g~10ml/g,进一步优选2ml/g~6ml/g,例如4ml/g。
[0022]
步骤a)中,所述的碱可以为有机碱或无机碱,所述的有机碱优选三乙胺、n,n-二异丙基乙胺和4-二甲氨基吡啶中的一种或多种,进一步优选三乙胺。所述的无机碱优选氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、碳酸氢钠和碳酸铯中的一种或多种。
[0023]
步骤a)中,所述的碱与所述的2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1的摩尔比值优选1~2,例如1。
[0024]
步骤a)中,所述的氨基保护试剂可以为乙酰氯、乙酸酐或二(叔丁基)二碳酸酯,优选二(叔丁基)二碳酸酯。
[0025]
步骤a)中,所述的氨基保护试剂与所述的2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1的摩尔比值优选1~2,例如1。
[0026]
步骤a)中,所述的缩合反应的温度优选0~40℃,进一步优选20℃~25℃。
[0027]
步骤a)中,所述的缩合反应的进程可以采用本领域常规检测方法(例如tlc、hplc或nmr)进行检测,一般以所述的2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1消失为反应的终点,所述的缩合反应的时间优选1小时~24小时,进一步优选2小时~12小时,例如10小时。
[0028]
步骤a)优选在保护气体保护下进行,所述的保护气体优选氮气和/或氩气。
[0029]
步骤a)优选采用以下后处理步骤:反应结束后,加入碱、萃取、洗涤、干燥、除去溶剂,得到纯化后的卡利拉嗪中间体2。所述的碱优选无机碱,所述的无机碱优选碳酸氢钠。所
述的碳酸氢钠的用量优选10ml~20ml饱和碳酸氢钠溶液。所述的洗涤优选采用水。所述的洗涤的次数优选1次~3次。所述的干燥优选采用无水硫酸钠或者无水硫酸镁。所述的除去溶剂优选减压除去溶剂。
[0030]
步骤a)优选采用以下步骤:向2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1、碱和溶剂形成的溶液中,加入氨基保护试剂与溶剂形成的溶液,进行缩合反应得到所述的卡利拉嗪中间体2即可。所述的“2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1、碱和溶剂形成的溶液”的温度优选5℃~10℃。所述的加入的方式优选滴加。所述的滴加的速度以维持反应体系温度不超过10℃为准。
[0031]
步骤a)进一步优选以下步骤:2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1与溶剂形成的溶液中,加入碱,降温至0-10℃(优选5℃~10℃),滴加氨基保护试剂,滴毕,升温至20-25℃,反应结束后,加入饱和碳酸钠溶液,分离相,用na2so4干燥,浓缩,得到卡利拉嗪中间体2。
[0032]
步骤b)中,所述的溶剂优选有机溶剂和水的混合溶剂。所述的有机溶剂优选醚类溶剂;所述的醚类溶剂优选四氢呋喃。所述的有机溶剂与所述的水的体积比值优选1~10,进一步优选2~6,例如4。
[0033]
步骤b)中,所述的溶剂与所述的卡利拉嗪中间体2的体积质量比值优选1ml/g~20ml/g,进一步优选10ml/g~15ml/g,例如12.5ml/g。
[0034]
步骤b)中,所述的碱优选无机碱;所述的无机碱优选氢氧化钠、氢氧化钾、氢氧化锂中的一种或多种,进一步优选氢氧化锂。所述的氢氧化锂可以为常规市售的一水合氢氧化锂试剂。
[0035]
步骤b)中,所述的碱与所述的卡利拉嗪中间体2的摩尔比值优选1.0~5.0,进一步优选1.1~3.0,例如2.0。
[0036]
步骤b)中,所述的水解反应的温度优选10℃~60℃,进一步优选20℃~50℃,例如40℃。
[0037]
步骤b)中,所述的水解反应的进程可以采用本领域常规检测方法(例如tlc、hplc或nmr)进行检测,一般以所述的卡利拉嗪中间体2消失时为反应的终点,所述的水解反应的时间优选1小时~12小时,进一步优选2小时~10小时,例如6小时。
[0038]
步骤b)中,所述的酸优选无机酸。所述的无机酸优选盐酸。所述的盐酸可以为常规市售盐酸试剂,所述的盐酸的浓度优选1mol/l~12mol/l,进一步优选2mol/l~10mol/l,例如6mol/l。
[0039]
步骤b)中,所述的酸与所述的卡利拉嗪中间体2的摩尔比值优选1~10,进一步优选2~6,例如5。
[0040]
步骤b)中,所述的酸化的温度优选-5℃~25℃,进一步优选0℃~5℃。
[0041]
步骤b)中,所述的酸化的进程可以采用本领域常规检测方法(例如tlc、hplc或nmr)进行检测,所述的酸化的时间优选0.5小时~5.0小时,进一步优选1.0小时~3.0小时,例如1.0小时。
[0042]
步骤c)中,所述的有机溶剂优选酮类溶剂、卤代烃类溶剂和醇类溶剂中的一种或多种。所述的酮类溶剂优选丙酮。所述的卤代烃类溶剂优选氯代烃类溶剂;所述的氯代烃类溶剂优选二氯甲烷;所述的醇类溶剂优选异丙醇。
[0043]
步骤c)中,所述的有机溶剂与所述的卡利拉嗪中间体3的体积质量比值优选1.0ml/g~20.0ml/g,进一步优选2.0ml/g~8.0ml/g,例如5.2ml/g、5.7ml/g或6.0ml/g。
[0044]
步骤c)中,所述的催化剂优选氯甲酸乙酯、氯甲酸异丁酯、n,n-羰基二咪唑、甲烷磺酰氯、对甲苯磺酰氯、对硝基苯磺酰氯、二碳酸二叔丁酯、二环己基碳二亚胺、二异丙基碳二亚胺、1-(3-二甲胺基丙基)-3-乙基碳二亚胺、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐、4-n,n-二甲基吡啶、1-羟基苯并三氮唑、4-吡咯烷基吡啶、o-(7-氮杂苯并三氮唑-1-基)-二(二甲胺基)碳鎓六氟磷酸盐、o-(苯并三氮唑-1-基)-二(二甲胺基)碳鎓六氟磷酸盐、o-(5-氯苯并三氮唑-1-基)-二(二甲胺基)碳鎓六氟磷酸盐、o-(苯并三氮唑-1-基)-二(二甲胺基)碳鎓四氟硼酸盐、o-(n-丁二酰亚胺基)-二(二甲胺基)碳鎓四氟硼酸盐、o-(n-endo-5-降莰烯-2,3-二碳二酰亚胺)-二(二甲胺基)碳鎓四氟硼酸盐、苯并三氮唑-1-基氧-三(二甲胺基)鏻鎓六氟磷酸盐、苯并三氮唑-1-基氧-三(四氢吡咯基)鏻鎓六氟磷酸盐、二苯基磷酰氯、氰代磷酸二乙酯、叠氮化磷酸二苯酯硫代二甲基磷酰基叠氮和二(2-氧-3-唑烷基)磷酰氯中的一种或多种,进一步优选n,n-羰基二咪唑、1-羟基苯并三唑和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐中的一种或多种。
[0045]
步骤c)中,所述的催化剂与所述的卡利拉嗪中间体3的摩尔比值优选1.0~2.0,进一步优选1.1~1.5,例如1.2。
[0046]
步骤c)中所述碱可以为有机碱和/或无机碱。所述的有机碱优选三乙胺、n,n-二异丙基乙胺和4-二甲氨基吡啶中的一种或多种,进一步优选三乙胺和/或n,n-二异丙基乙胺。所述的无机碱优选氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、碳酸氢钠和碳酸铯中的一种或多种。
[0047]
步骤c)中,所述的碱与所述的卡利拉嗪中间体3的摩尔比值优选1.0~3.0,进一步优选1.0~2.0,例如1.0或1.1。
[0048]
步骤c)中,所述的1-(2,3-二氯苯基)-哌嗪4与所述的卡利拉嗪中间体3的摩尔比值优选1.0~3.0,进一步优选1.0~2.0,例如1.0。
[0049]
步骤c)中,所述的缩合反应的温度优选0~40℃,进一步优选10℃~30℃;再进一步优选20℃~25℃。
[0050]
步骤c)中,所述的缩合反应的进程可以采用本领域常规检测方法(例如tlc、hplc或nmr)进行检测,一般以所述的卡利拉嗪中间体3消失为反应的终点,所述的缩合反应的时间优选1小时~24小时,进一步优选6小时~12小时,例如12小时。
[0051]
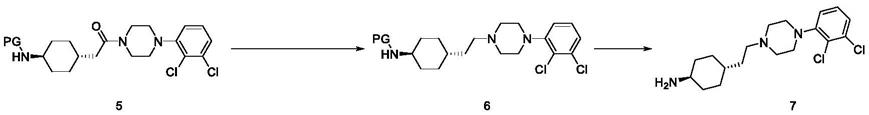
本发明还提供了一种卡利拉嗪中间体7的制备方法,其特征在于:按照上述方法制得卡利拉嗪中间体5之后,还包括以下步骤:
[0052]
d)有机溶剂中,将所述的卡利拉嗪中间体5溶于与还原剂进行还原反应,得到卡利拉嗪中间体6,然后再与酸进行脱除氨基保护基的反应,得到卡利拉嗪中间体7即可;
[0053][0054]
其中,pg为氨基保护基或氢。所述的氨基保护基优选叔丁氧羰基或乙酰基等。
[0055]
步骤d)中,所述的有机溶剂优选醚类溶剂;所述的醚类溶剂优选四氢呋喃。
[0056]
步骤d)中,所述的有机溶剂与所述的卡利拉嗪中间体5的体积质量比值优选
1.0ml/g~20.0ml/g,进一步优选2.0ml/g~8.0ml/g,例如6.0ml/g。
[0057]
步骤d)中,所述还原剂可以为三氟化硼乙醚和/或硼氢化钠。
[0058]
步骤d)中,所述的还原剂与所述的卡利拉嗪中间体5的摩尔比值优选1.0~5.0,进一步优选1.5~3.0,例如1.9或2.9。
[0059]
步骤d)中,所述的还原反应的温度优选0~25℃,进一步优选0℃~10℃,例如0℃~5℃。
[0060]
步骤d)中,所述的还原反应的进程可以采用本领域常规检测方法(例如tlc、hplc或nmr)进行检测,一般以所述的卡利拉嗪中间体5消失为反应的终点,所述的还原反应的时间优选0.5小时~24小时,进一步优选2小时~10小时,例如5小时。
[0061]
步骤d)中,所述的酸优选无机酸。所述的无机酸优选盐酸和/或硫酸。所述的盐酸可以为常规市售盐酸试剂;所述的盐酸的摩尔浓度优选1mol/l~12mol/l,进一步优选2mol/l~6mol/l,例如4mol/l。所述的硫酸可以为常规市售硫酸试剂;所述的硫酸的摩尔浓度优选1mol/l~18mol/l。
[0062]
步骤d)中,所述的酸与所述的卡利拉嗪中间体5的摩尔比值优选1~20,进一步优选2~15,例如8。
[0063]
步骤d)中,所述的脱除氨基保护基的反应的温度优选10℃~80℃,进一步优选20℃~60℃,例如50℃。
[0064]
步骤d)中,所述的脱除氨基保护基的反应的进程可以采用本领域常规检测方法(例如tlc、hplc或nmr)进行检测,一般以所述的卡利拉嗪中间体5消失为反应的终点,所述的脱除氨基保护基的反应的时间优选1.0小时~10.0小时,进一步优选2.0小时~6.0小时,例如3.0小时。
[0065]
本发明还提供了一种卡利拉嗪的制备方法,其特征在于:按照上述方法制得卡利拉嗪中间体7之后,还包括以下步骤:
[0066]
e)有机溶剂中,将步骤d)得到的卡利拉嗪中间体7与碱和二甲基-氨基甲酰氯,进行缩合反应得到卡利拉嗪(反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己基}-脲)即可;
[0067][0068]
步骤e)中,所述的有机溶剂优选卤代烃类溶剂;所述的卤代烃类溶剂优选氯代烃类溶剂;所述的氯代烃类溶剂优选二氯甲烷。
[0069]
步骤e)中,所述的有机溶剂与所述的卡利拉嗪中间体7的体积质量比值优选1.0ml/g~20.0ml/g,进一步优选2.0ml/g~8.0ml/g,例如6.0ml/g。
[0070]
步骤e)中,所述的碱优选有机碱或无机碱;所述的有机碱优选三乙胺、n,n-二异丙基乙胺和4-二甲氨基吡啶中的一种或多种,进一步优选三乙胺。所述的无机碱优选氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、碳酸氢钠和碳酸铯中的一种或多种。
[0071]
步骤e)中,所述的碱与所述的卡利拉嗪中间体7的摩尔比值优选1.0~5.0,进一步优选1.5~3.0,例如2.0。
[0072]
步骤e)中,所述的二甲基-氨基甲酰氯与所述的卡利拉嗪中间体7的摩尔比值优选
1.0~2.0,进一步优选1.0~1.5,例如1.0。
[0073]
步骤e)中,所述的缩合反应的温度优选0~40℃,进一步优选20℃~30℃,例如25℃。
[0074]
步骤e)中,所述的缩合反应的进程可以采用本领域常规检测方法(例如tlc、hplc或nmr)进行检测,一般以所述的卡利拉嗪中间体7消失为反应的终点,所述的缩合反应的时间优选2小时~24小时,进一步优选6小时~12小时,例如10小时。
[0075]
本发明中,所述的卡利拉嗪的制备方法优选采用以下合成路线:
[0076]
本发明所用试剂和原料均市售可得。
[0077]
本发明的积极进步效果在于:原料易得,操作简便,中间体及产物容易纯化,有效控制成品质量,保证药物质量安全可控,反应收率高,适合于工业化生产。
具体实施方式
[0078]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0079]
实施例1反式2-{1-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙酸乙酯(卡利拉嗪中间体2)的制备
[0080]
于50ml三口瓶中加入4g(0.018mol)反式2-[1-(4-氨基]-环己基)-乙酸乙酯盐酸盐1和16ml二氯甲烷,然后加入1.8g(0.018mol)的三乙胺,混合物冷却至5-10℃,然后在氮气保护下,滴加4.0g(0.018mol)的二(叔丁基)二碳酸酯的10ml二氯甲烷溶液,滴加完后20℃搅拌,10h后,反应结束,加入20ml饱和碳酸氢钠溶液,分离相。用10ml的水洗涤有机层,分离相,用无水na2so4干燥有机相,减压除去溶剂,30℃下减压烘干,得4.64g的目标化合物,产率90.0%。hplc纯度98.5%。
[0081]
实施例2反式2-{1-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙酸(卡利拉嗪中间体3)的制备
[0082]
于50ml三口瓶中加入4.0g(0.014mol)反式2-{1-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙酸乙酯(卡利拉嗪中间体2)、40ml四氢呋喃、10ml蒸馏水和1.18g(0.028mol)一水
合氢氧化锂,40℃搅拌反应6h,反应结束后,减压除去四氢呋喃,加入14ml(6m)的盐酸酸化,冰浴搅拌1h后,过滤,10ml蒸馏水洗涤,50℃减压干燥烘干,得1.8g目标化合物,产率90.0%,hplc纯度99.0%。
[0083]
实施例3叔丁基(1r,4r-4-(2-(4-(2,3-二氯苯基)哌嗪-1-基)-2-氧乙基)环己基)氨基甲酸酯(卡利拉嗪中间体5)的制备
[0084]
方法a:于50ml三口瓶中,加入3.86g(0.015mol)反式2-{1-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙酸(卡利拉嗪中间体3)、20ml丙酮和2.9g(0.018mol)n,n-羰基二咪唑,25℃搅拌4h,加入2ml异丙醇,25℃搅拌1h,随后加入2.1ml三乙胺和3.47g(0.015mol)1-(2,3-二氯苯基)-哌嗪4,室温搅拌12小时,反应结束后,加入80ml蒸馏水,搅拌1h,过滤,40ml蒸馏水洗涤。在45℃减压烘干得6.4g目标化合物,收率90.0%,hplc纯度99.5%。
[0085]
方法b:于50ml三口瓶中加入5g(0.019mol)反式2-{1-[4-(n-叔丁氧基羰基)-氨基]-环己基}-乙酸(卡利拉嗪中间体3)、30ml二氯甲烷、4.5g(0.019mol)1-(2,3-二氯苯基)-哌嗪4、2.46g(0.021mol)n,n-二异丙基乙胺,降温至0℃,搅拌,加入2.89g(0.021mol)1-羟基苯并三唑和3.4g(0.0177mol)1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐,0℃搅拌1h后,25℃搅拌12h,反应结束后,加入30ml质量浓度10%的盐酸(所述的质量浓度是指氯化氢的质量占盐酸溶液总质量的百分比),分离相,10ml饱和碳酸氢钠洗涤,分离相,加入50ml饱和氯化钠溶液,分离相,无水硫酸钠干燥,减压除去二氯甲烷,得到8g目标化合物,收率83.0%,hplc纯度99.3%。
[0086]
实施例4叔丁基((1r,4r)-4-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)环己基)氨基甲酸酯(卡利拉嗪中间体6)的制备
[0087]
于100ml三口瓶中加入5g(0.011mol)叔丁基(1r,4r-4-(2-(4-(2,3-二氯苯基)哌嗪-1-基)-2-氧乙基)环己基)氨基甲酸酯和30ml四氢呋喃,0-5℃搅拌,加入2g(0.021mol)硼氢化钠和4.53g(0.032mol)三氟化硼乙醚,0-5℃搅拌1h后,室温搅拌5小时,反应结束后,加入20ml水,升温至回流,反应结束后,减压除去四氢呋喃,固体过滤,叔丁醇重结晶,得4g目标化合物,收率82.0%,hplc纯度99.3%。
[0088]
实施例5(1r,4r)-4-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)环己烷-1-胺盐酸盐(卡利拉嗪中间体7)的制备
[0089]
于100ml三口瓶中加入5g(0.011mol)叔丁基((1r,4r)-4-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)环己基)氨基甲酸酯和15ml乙醇,室温搅拌,加入22ml盐酸(4mol/l),加热至50℃,反应3h后,减压除去乙醇,冷却至0-5℃,抽滤,洗涤,40℃减压干燥,得4.1g目标化合物,收率95.0%。
[0090]
实施例6反式-n-{4-[2-[4-(2,3-二氯苯基)哌嗪-1-基]乙基]环己基}-n',n'-二甲基脲(卡利拉嗪)的制备
[0091]
于50ml三口瓶中加入5g(0.013mol)(1r,4r)-4-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)环己烷-1-胺盐酸盐(卡利拉嗪中间体7)、30ml二氯甲烷和2.58g(0.026mol)三乙胺,0-5℃搅拌,滴加1.37g(0.013mol)二甲基-氨基甲酰氯的5ml二氯甲烷溶液,滴加完毕后25℃反应10h,反应结束后,加入30ml饱和氯化钠溶液,分离相,无水硫酸钠干燥,减压除去二氯甲烷,甲醇重结晶,得4.5g目标化合物,收率83.0%,hplc纯度99.5%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。