1.本发明属于基因测序领域,具体涉及一种新型三代测序方法。
背景技术:
2.随着单细胞或空间转录组测序技术的发展,结合单细胞信息可以从细胞角度探究细胞异质性及细胞间差异,结合空间位置信息则可以深入理解组织微环境和细胞间的相互作用。目前成熟的单细胞或空间转录组测序手段都是将测序分子 (cdna)两端接上区分单个细胞的条形码(cb,cell barcode)或区分空间信息的条形码(sb,spatial barcode),以及每条cdna都带有唯一分子标识符(umi, unique molecular identifier),接下来再通过下一代测序技术(ngs,nextgeneration sequencing)打断cdna后进行高通量测序获得序列信息,最终再通过cb或sb区分出单个细胞或单个空间位置的序列信息。因此,获得准确的sb (或cb)和umi对于空间(或单细胞)转录组测序至关重要。
3.但ngs技术因其必须将cdna打断的特性,无法获得序列全长信息,限制了区分同一个基因的mrna因选择性剪接(as,alternative splicing)产生的多种异构体(isoforms),不利于mrna转录后水平调控和转录本多态性的研究。目前高通量测序平台中,仅有作为第三代测序技术的牛津纳米孔测序技术(ont, oxford nanopore technologies)和太平洋生物科学公司(pacbio,pacificbiosciences)开发的单分子荧光测序(smrt,single molecule real timesequencing)两大测序平台可以对全长cdna实现端到端(end-to-end)的完整测序。pacbio的iso-seq工作流程作为全长cdna测序的金标准,可以在一次运行周期内产生约二十万条高质量的全长cdna读长(reads)。而ont目前能以一张minion芯片产出百万级别的reads数目,其强大的灵活性(flexibility)、可扩展性(scalability)和更低的成本,已经越来越受到各实验室青睐,成为全长转录组研究的首选工具。
4.然而,ont测序因纳米孔读取电信号的测序原理决定了其测序准确度相对较低,错误率可达5~25%,特别是同聚物(homopolymer)和串联重复区域(tandemrepeats region)的错误。如果需要对单细胞或空间转录组文库(library)进行全长 cdna测序,会导致cb或sb无法被准确拆解,能正确识别的单细胞或空间位置的reads很少。
5.为解决这一问题,volden等最早于2018年提出r2c2方法(rolling circleamplification to concatemeric consensus),即通过重组一段200bp的无关序列环化cdna后利用滚环扩增(rca,rolling circle amplification)生成一条长的串联重复序列(concatemer)并进行ont测序,后将被多次测到的cdna从其串联重复序列拆解后进行相互矫正成共识序列(consensus),以提高测序准确度。 volden等又在2020年将r2c2方法应用于商品化10
×
genomic单细胞转录组平台(10xr2c2),但其拆解出sb的reads仅占所有reads的~32.75%,而同时拆解出sb和umi的匹配率为~15.86%。除此之外,类似于r2c2但应用场景不同的方法也在近几年不断被开发出来,例如针对超短dna的hifre、针对 phospho-rna的circaid-p-seq、针对循环肿瘤dna(ctdna,circulating tumordna)的tp53-specific cyclomicsseq等方法,证明通过串联重复序列矫正ont 的理论到了广泛应用。
6.与此同时,另一种提高条形码和umi匹配率的方法也被提出。scnaumi-seq 和scnapbar等方法是对同一单细胞测序文库分别进行了二代和ont测序,以二代数据为参考指导,识别拆解ont数据中的cb和umi,以此获得准确cb 和umi的ont reads。但是该类方法只能提高条形码和umi的匹配率,并不能提高cdna的测序准确率,而且其拆解出cb的reads仅占所有reads的~38.76%,同时拆解出cb和umi的匹配率为~29.45%,并不能真正解决条形码和umi匹配率不高的问题。
技术实现要素:
7.有鉴于此,本发明提供了一种核酸测序方法,所述核酸测序方法包括:s1将待测核酸序列环化,得到环化核酸序列,所述待测核酸序列包括核酸内嵌序列、标识序列以及分别位于所述待测核酸序列两端的固定序列,所述环化的环化方式包括plp;s2以所述环化核酸序列作为模板进行滚环扩增,得到滚环扩增产物; s3对所述滚环扩增产物进行测序,获得原始串联重复序列;s4基于所述固定序列,对所述原始串联重复序列进行第一轮标识匹配,提取标识区域第一序列; s5基于所述固定序列和所述标识序列,对所述原始串联重复序列进行第二轮标识匹配,从而对所述标识区域第一序列进行校正,得到标识区域第二序列;s6基于所述标识区域第二序列,对所述原始串联重复序列进行拆解,得到一个或多个亚读长,所述亚读长包括所述核酸内嵌序列的中间产物和所述标识区域第二序列; s7对所述亚读长进行多序列比对,提取核酸第一共识序列,所述核酸第一共识序列即视为所述核酸内嵌序列的测序结果。
8.进一步地,所述方法进一步包括:在所述第二轮标识匹配前,基于所述标识区域第一序列,对所述原始串联重复序列进行比对,提取核酸第二共识序列。
9.进一步地,所述标识序列包括唯一分子标识符(umi)和/或条形码,其中,所述待测核酸序列的唯一分子标识符是唯一确定的,所述待测核酸序列的条形码提供用于鉴别所述待测核酸序列的信息。
10.进一步地,所述条形码包括空间条形码和/或细胞条形码。
11.进一步地,所述标识区域第一序列包括与所述固定序列相匹配的固定序列匹配序列,以及在所述标识序列所在方向上与所述固定序列匹配序列直接相连的 28-58bp。
12.进一步地,所述s3进一步包括对所述滚环扩增产物进行第三代测序并获得所述滚环扩增产物的第三代测序数据。
13.进一步地,所述第三代测序包括纳米孔测序和/或单分子荧光测序。
14.进一步地,所述s5进一步包括对所述待测核酸序列进行第二代测序并获得所述待测核酸序列的第二代测序数据。
15.进一步地,所述第二代测序包括离子半导体测序、焦磷酸测序、连接测序、合成测序、组合探针锚点合成测序和可逆性末端终止测序中的一种或多种。
16.进一步地,所述校正为利用参考序列对所述标识区域第一序列进行校正,其中,所述参考序列来自所述待测核酸序列的第二代测序数据。
17.进一步地,所述s1进一步包括扩增所述待测核酸序列以生成所述核酸内嵌序列的文库,其中,所述扩增先于所述将待测核酸序列环化。
18.进一步地,所述核酸内嵌序列包括dna。
19.进一步地,所述核酸内嵌序列包括生物样本中的rna逆转录形成的cdna。
20.进一步地,所述核酸第一共识序列为高精度。
21.进一步地,按照引导树算法对所述原始串联重复序列进行比对。
22.进一步地,所述比对可选地为profile-to-profile比对。
23.进一步地,按照动态规划算法对所述亚读长进行多序列比对。
24.进一步地,所述生物样本包括新鲜组织切片、新鲜单细胞悬液和提取的组织 rna中的一种或多种。
25.进一步地,所述生物样本源自生物体的组织和/或器官。
26.进一步地,所述生物样本包括心脏、肾脏、肝脏、脾脏、胃、肺、卵巢、乳房、淋巴结、舌、脑、大肠、小肠、眼睛、骨骼肌、睾丸、甲状腺样品中的一种或多种。
27.另一方面,本发明还提供了一种三代测序系统,其特征在于,包括:序列信息接收模块,用于接收待测核酸序列的原始串联重复序列信息,其中,所述待测核酸序列包括核酸内嵌序列、标识序列以及分别位于所述待测核酸序列两端的固定序列,所述原始串联重复序列是以环化的所述待测核酸序列为模板,滚环扩增得到;第一匹配模块,用于基于所述固定序列,对所述原始串联重复序列进行第一轮标识匹配,提取标识区域第一序列;第二匹配模块,用于基于所述固定序列和所述标识序列,对所述原始串联重复序列进行第二轮标识匹配,从而对所述标识区域第一序列进行校正,得到标识区域第二序列;拆解模块,用于基于所述标识区域第二序列,对所述原始串联重复序列进行拆解,得到一个或多个亚读长,所述亚读长包括所述核酸内嵌序列和所述标识区域第二序列;比对模块,用于对所述亚读长进行多序列比对,提取核酸第一共识序列,所述核酸第一共识序列即视为所述核酸内嵌序列的测序结果。
28.进一步地,所述环化的环化方式包括plp。
29.所述比对模块还可以用于:在所述第二轮标识匹配前,基于所述标识区域第一序列,对所述原始串联重复序列进行比对,提取核酸第二共识序列。
30.进一步地,所述标识序列包括唯一分子标识符(umi)和/或条形码,其中,所述待测核酸序列的唯一分子标识符是唯一确定的,所述待测核酸序列的条形码提供用于鉴别所述待测核酸序列的信息。
31.进一步地,所述条形码包括空间条形码和/或细胞条形码。
32.进一步地,所述标识区域第一序列包括与所述固定序列相匹配的固定序列匹配序列,以及在所述标识序列所在方向上与所述固定序列匹配序列直接相连的 28-58bp。
33.进一步地,所述待测核酸序列的原始串联重复序列信息是通过第三代测序得到。
34.进一步地,所述第三代测序包括纳米孔测序和/或单分子荧光测序。
35.进一步地,所述序列信息接收模块还可以用于接收所述待测核酸序列的第二代测序信息。
36.进一步地,所述第二代测序包括离子半导体测序、焦磷酸测序、连接测序、合成测序、组合探针锚点合成测序和可逆性末端终止测序中的一种或多种。
37.进一步地,所述校正为利用参考序列对所述标识区域第一序列进行校正,其中,所述参考序列来自所述待测核酸序列的第二代测序信息。
38.进一步地,所述核酸内嵌序列包括dna。
spatial gene expression,10x genomics公司)的引发测序的“read1”和用于模板转换的“模板转换寡核苷酸(tso)”。根据实际需求,固定序列还可以是基于其他测序平台的接头序列。
52.术语“扩增”通常是指将目标核酸或其部分形成一个或多个拷贝的任意过程。术语“滚环扩增(rca)”是指,由滚环机制扩增环状核酸模板(例如,dna 环)的核酸扩增反应;滚环扩增反应通过引物杂交至所述环状核酸模板而启动,核酸聚合酶然后延伸引物,所述引物不断被杂交至所述环状核酸模板,以反复不断地复制所述环状核酸模板的序列。所述滚环扩增反应通常产生包含环状核酸模板序列的串联重复单元的串联重复序列(concatamer)。在一些实施例中,可以通过各种环化方式(例如,plp、r2c2、bta、cl、sl),将“待测核酸序列”形成相应的用于所述滚环扩增反应的所述环状核酸模板。
53.如本文所使用,“标识序列”是指与所述期望测序的核酸序列相连、用于鉴定待测核酸序列的核苷酸序列。如本文所使用,“标识序列”包括umi和/或条形码,所述标识序列的一端与“固定序列”相连,另一端与“期望测序的核酸序列”相连。在一些实施例中,“umi”可用于从不同的扩增产物中区分出同一种扩增产物(例如,每个被扩增的所述期望测序的核酸序列都会有一个不同的随机 umi,相同的umi代表扩增产物来自相同的所述期望测序的核酸序列)。如本文所使用,“条形码”包括细胞条形码和空间条形码。在一些实施例中,空间条形码可以包含提供与条形码相关联的所述期望测序的核酸序列的空间位置的信息的核酸序列,例如样品的坐标。在一些实施例中,细胞条形码可以包含提供用于确定哪种所述期望测序的核酸序列源自哪个细胞的信息的核酸序列。
54.如本文所使用,“标识区域第一序列”是指,原始串联重复序列中,与所述固定序列匹配的序列,及在标识序列方向上与所述固定序列匹配的序列直接相连的28-58bp;“标识区域第二序列”是指,利用“待测核酸序列”的二代测序数据中的“固定序列”和“标识序列”的序列信息(本发明提供的方法将其视为正确的序列信息),对原始串联重复序列中的“标识区域第一序列”进行校正后得到的对“标识序列”的测序结果。
55.如本文所使用,“样品”可以包含单细胞、多个细胞、组织、器官或微生物等。“样品”还可以是源自含单细胞、多个细胞、组织、器官或微生物等的dna 或rna。
56.如本文所使用,“cdna(互补dna)”是指一段rna(通常为mrna,但不限于mrna)经逆转录形成的dna。“全长cdna”是指,至少包括作为来源的mrna的完整编码区的cdna。
57.如本文所使用,“核酸内嵌序列的中间产物”是指串联重复序列的环状核酸模板序列的串联重复单元中“核酸内嵌序列”对应的序列。鉴于滚环扩增反应引起的误差,所述核酸内嵌序列的中间产物可以与“核酸内嵌序列”一致,也可以与“核酸内嵌序列”的部分一致。
附图说明
58.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍。显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。
59.图1为本发明方法的工作流程图;
60.图2为五种自成环方法的比较图;
61.图3为本发明方法的性能效果图;
62.图4为本发明方法提升异构体(isoforms)准确度的实验图;
63.图5为本发明方法促进新型剪接点的识别的实验图;
64.图6为本发明方法促进rna编辑位点的定量的实验图;
65.图7为本发明提供的一种核酸测序方法的流程图;
66.图8为本发明提供的三代测序系统的功能模块图。
具体实施方式
67.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
68.需要说明的是,在本文中,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者装置不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者装置所固有的要素。在没有更多限制的情况下,由语句“包括一个
……”
限定的要素,并不排除在包括该要素的过程、方法、物品或者装置中还存在另外的相同要素。
69.如在本说明书中使用的,术语“大约”,典型地表示为所述值的 /-5%,更典型的是所述值的 /-4%,更典型的是所述值的 /-3%,更典型的是所述值的 /-2%,甚至更典型的是所述值的 /-1%,甚至更典型的是所述值的 /-0.5%。
70.在本说明书中,某些实施方式可能以一种处于某个范围的格式公开。应该理解,这种“处于某个范围”的描述仅仅是为了方便和简洁,且不应该被解释为对所公开范围的僵化限制。因此,范围的描述应该被认为是已经具体地公开了所有可能的子范围以及在此范围内的独立数字值。例如,范围的描述应该被看作已经具体地公开了子范围如从1到3,从1到4,从1到5,从2到4,从2 到6,从3到6等,以及此范围内的单独数字,例如1,2,3,4,5和6。无论该范围的广度如何,均适用以上规则。
71.实施例一:材料与方法
72.1.1夹板设计及合成
73.本发明中plp(padlockprobe)环化方式(以下简称为plp)的夹板设计可根据不同cdna文库两端的固定序列灵活设计,主要是以能够通过夹板将cdna 文库两端首尾相接为目的,通常需要设计反向互补的两条夹板,以实现cdna 正反链的各自环化。以空间转录组平台(visium spatial gene expression,10xgenomics公司)流程产生的全长cdna为例,其两端固定序列为:read1:5
’ꢀ‑
ctacacgacgctcttccgatct-3’;tso:5
’ꢀ‑
aagcagtggtatcaacgcagagtacatggg-3’。则反向互补的两条夹板序列分别为:splint-top: 5
’‑
agatcggaagagcgtcgtgtagaagcagtggtatcaacgcagagtaca tggg-3’;splint-bottom: 5
’‑
cccatgtactctgcgttgataccactgcttctacacgacgctcttccga tct-3’。
74.1.2全长文库的磷酸化扩增及产物纯化
75.1.2.1全长文库磷酸化扩增
76.按表1扩增cdna,并按表2循环设置pcr仪。
77.表1 cdna扩增体系备注:由于kapahifi热启动dna聚合酶的3
’‑5’
核酸外切酶活性不是热启动的,为防止配制过程中cdna模板或引物被降解,应在冰上配制体系。
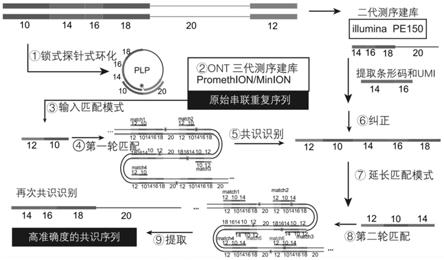
78.表2 pcr仪的设置
79.1.2.2产物纯化及质检
80.使用0.6
×
的ampure xp磁珠对扩增产物进行纯化,纯化后溶解于20μl无核酸酶水。使用qubit仪器测定产物浓度,使用qseq仪器或其他核酸质检仪质检产物长度,浓度大于20ng/ul、长度与扩增前一致(一般大于1kb)即可。
81.1.3滚环扩增及产物纯化
82.1.3.1cdna环化
83.图1中各编号所表示的含义如下:
84.10:read1,12:tso,14:条形码,16:poly(dt)vn,18:umi,20:核酸内嵌序列。
85.如图1步骤
①
所示,取100ng全长cdna,以qsep1测得的片段长度峰值为平均片段长度,根据下述公式计算摩尔浓度,使用与cdna等摩尔数的夹板 (splint-top和splint-bottom)进行混合,用无核酸酶水稀释至10μl。pcr仪上 95℃孵育2min,立即放到冰上冷却。
[0086][0087]
按表3配制连接反应液。室温连接1h后,加入2μl exo iii:lambda exo: exo i=1:9:10比例的混合酶,加热块上37℃孵育30min后使用0.6
×
的ampurexp磁珠对环化产物进行纯化,纯化后溶解于50μl无核酸酶水。
[0088]
表3连接反应液配置体系
[0089]
1.3.2滚环扩增
[0090]
使用3’端最后两位磷酸二酯键进行过硫代磷酸酯改性的耐3
’‑5’
外切酶消化的随机六聚体作为引物在环状dna上引发扩增。使用equi phi29 dna聚合酶作为扩增酶,由于其链置换特性,当围绕环状模板延伸一圈后,与自身新合成链5’端接触时,能将5’端置换,继续合成新链,从而实现单链环状模板的串联重复扩增。并且,新合成的单链上仍能结合随机引物,引发其反向互补链的合成。
[0091]
将环化产物各分为5管(每管约10μl),按表4配制。
[0092]
表4
[0093]
加热块上95℃变性2min后立即放到冰上,并加入如表5试剂。pcr仪上 42℃孵育2h,后37℃孵育过夜或8h。
[0094]
表5
[0095]
1.3.3去分支化(debranching)
[0096]
随机引物引发的扩增是多扩增位点并行的,导致扩增产物为分支状单双链杂化复合物,可用t7核酸内切酶(endonuclease)i这样的能识别分枝形dna并切割的去分支化内切酶。
[0097]
由于扩增产物为粘稠的长片段dna,小心将同一样本的5个扩增管合并到同一个低粘附1.5ml离心管中并补无核酸酶水至300μl,加入0.5
×
spriselect 磁珠,轻弹管底混匀。试管混匀器上混匀5min,放到磁力架上静置2min后吸去上清,80%乙醇清洗两次,晾干后加入90μl无核酸酶水重悬磁珠,并加入10 μlnebuffer 2和5μl t7e1,轻弹管底混匀后连同试管混匀器放到37℃孵箱中混匀孵育2-3h,视成团磁珠消散情况停止反应。
[0098]
将离心管瞬离后放到磁力架上静置2min,转移上清至新的1.5ml低粘附离心管,加入0.5
×
spriselect磁珠,轻弹管底混匀,纯化步骤如上所述,溶解于 15μl无核酸酶水。取1μl用9μl无核酸酶水稀释后qubit测定浓度,剩下的稀释后的样本可加2μl 6
×
loading buffer进行0.8%琼脂糖凝胶电泳。浓度大于 100ng/ul、长度大于10kb即可。
[0099]
1.4ont建库、上机测序及数据前处理
[0100]
根据ont连接测序试剂盒(sqk-lsk109)的标准流程对滚环扩增产物进行文库制备(如图1步骤
②
所示),在dna修复、末端修复并加a、连接无扩增条形码( barcode)后根据nebiocalculator将样品以100fmol的最终量混合,最后连接测序接头,并配置上机混合液。
[0101]
接下来,将上机混合液加载至质检合格的ont测序芯片中,设置 minknow软件中的测序参数,测序持续48h以上。使用guppy对测序得到的电信号(fast5格式)进行碱基拆解,得到原始串联重复序列(raw concatemerreads)(若minknow中采用“high basecalling”及以上的碱基识别模式则无需此步骤)。
[0102]
1.5高精度的共识序列(consensus read)生成(以下简称为本发明方法)
[0103]
首先使用r包biostrings(https://bioconductor.org/packages/biostrings)中的函数matchpattern,以“固定序列(tso read1)”为模式(即输入匹配模式) 对原始串联重复序列(raw concatemer reads)进行第一轮匹配,考虑到原始reads 的高错误率,该匹配模式允许一定的碱基插入、缺失和错配(如图1步骤
③
所示)。若某条reads匹配成功,则从匹配区域沿标识序列方向截取28-58bp(优选为35 bp),以从原始串联重复序列(raw concatemer reads)中提取所有潜在的条形码和umi序列(如图1步骤
④
所示)。其中,“poly(dt)vn”起定位作用,即可以从所截取到的片段中是否含有部分“poly(dt)vn”序列(即重复的t碱基) 来判断是否将“条形码”和“umi”完整截取。
[0104]
接下来,可选地,使用r包decipher[12]中的函数alignseqs,对多个未比对序列按照引导树算法进行profile-to-profile比对,并从多序列比对结果中提取共识序列(consensus read)(如图1步骤
⑤
所示)。同时,对同一cdna,进行ngs建库和测序,以二代数据为“标准”序列,以辅助纠正从每条原始串联重复序列中共识识别(consensus calling)得到的条形码和umi序列,以得到正确的条形码和umi(如图1步骤
⑥
所示)。接着,以“固定序列 条形码序列 (tso read1 barcode)”为模式(即延长匹配模式),对原始串联重复序列再次使用函数matchpattern进行第二轮匹配(如图1步骤
⑦
所示)。与第一轮匹配相比,因“固定序列 条形码序列”模式的延长而提高了容错率,匹配数更多,共识识别也将更准确。并且使用第二轮匹配的区域,将原始串联重复序列拆解为亚读长(subreads)(如图1步骤
⑧
所示)。最后使用alignseqs对所述亚读长进行多序列比对,并将共识序列提取为高精度的cdna序列(即再次共识识别,如图1步骤
⑨
所示)。至此,将原始串联重复序列生成为正确的条形码和umi 以及高精度的cdna序列。
[0105]
在本发明的一些实施例中,新型三代测序方法如图7所示。
[0106]
实施例二:五种环化方式的比较
[0107]
本实施例首先系统性地将plp与其他四种全长cdna自成环方法进行了比较(如图2a所示),包括r2c2[参考文献6]以及pacbio测序中采用的茎环结构连接(sl)、inc-seq[参考文献13]中使用blunt/ta连接酶的双链自成环(bta) 以及circligase连接酶介导的单链自成环(cl)。
[0108]
本实施例统一将长为597bp的标准序列作为起始cdna,分别以各自成环方法的实验流程进行环化,接下来以r2c2方法的后续流程产生原始串联重复序列,通过c3poa软件(https://github.com/rvolden/c3poa)生成共识序列后,通过minimap2软件(https://
github.com/lh3/minimap2)与标准序列进行比对。最后通过共识序列的生成效率(consensus%)、比对率(mapped%)、比对长度(mappedlength)、缺失突变率(indels%)和碱基错配率(mismatch%)综合评估这五种方法。
[0109]
结果表明,plp和r2c2产生了最大数量的原始序列,并获得了最高的共识序列生成效率,分别为12.4%和30.9%;将五种方法生成的共识序列与标准序列比对后,bta和plp的比对率分别为99.5%和99.3%,其次是sl(98.5%)、 r2c2(98%)和cl(78.2%)(图2b)。如图2c所示,在比对长度的比较中,bta 和plp的比对长度显著更接近标准序列的长度,具体表现为:比对长度集中分布于相应小提琴图的顶端(宽度最大),中位数所对应的比对长度明显大于其他三者;而r2c2、cl和sl的长度明显更短(小提琴图中的实心点表示中位数,宽度表示频率)。在共识序列准确性(还可以理解为“精度”)的比较中,plp 的缺失突变率(图2d)和碱基错配率(图2e)最低,具体表现为:在相应小提琴图中,中位数所对应的缺失突变率和碱基错配率最低,且集中分布于小提琴图的底部(宽度最大)(小提琴图中的实心点表示中位数,宽度表示频率)。而在 plp、r2c2、bta、cl和sl这五种环化方式中,bta、cl和sl三种环化方式没有引入夹板序列来介导环化过程,属于随机成环,因此在实际应用场景中存在一定的偶然性。总的来看,plp在生成具有所需长度的高百分比共识序列和低百分比缺失突变和碱基错配方面表现出最高的效率。
[0110]
实施例三:本发明方法与已发表数据的比较
[0111]
本实施例着重于对10xr2c2和scnaumi-seq文章中阐明的数据进行比较,以各过程产生的reads与原始reads数目(basecalled reads)的比率进行比较,以探究各技术的效率。
[0112]
将原始串联重复序列矫正为共识序列后,如表6所示,在两端都具有接头序列(也可以理解为“固定序列”)的reads数占原始reads数的比率(both-endreads%)的比较中,本发明方法(以plp环化方式)的7个重复数据均能产生大于60%的reads,但scnaumi-seq中8个重复数据能产生平均57%的reads,而 10xr2c2的2个重复数据只能产生约32.1%和33.4%的reads。接下来,在以具有接头序列的基础上,比较了匹配正确条形码的reads数占原始reads数的比率 (both-end barcode reads%),本发明方法因工作原理是找到两端接头的同时匹配正确的条形码,因此比率与上一步(即匹配正确条形码的reads数占原始reads 数的比率)相比没有变化,而scnaumi-seq的8个重复数据下降至平均38.76%,10xr2c2的2个重复数据分别下降至21.66%和21.63%。最后,在以具有接头序列、正确条形码的基础上,比较了匹配正确umi的reads数占原始reads数的比率(both-end barcode&umi reads%),本发明方法的7个重复数据能产生平均 36.43%的reads,而scnaumi-seq中8个重复数据为平均29.45%,10xr2c2的2 个重复数据为16.10%与15.83%。
[0113]
表6各过程的reads数目比较
[0114]
除此之外,本实施例还以相同cdna样本与r2c2方法进行了比较。亚读长覆盖率(subread coverage)表示一条串联重复序列中所含的亚读长的数量。如图3a所示,与r2c2方法相比,本发明方法(以plp方式环化)具有大于10 条的亚读长覆盖率且比r2c2高大约30%,这表明本发明方法的滚环扩增的循环次数显著大于r2c2,进而亚读长之间的相互校正的成功率更高。接下来,以相同cdna样本进行ont测序,并运行scnapbar[参考文献11]的数据处理流程,只有18.5%的reads可以匹配正确的条形码。理论上,本发明方法(以plp环化方式)的夹板序列未引入无关序列,而r2c2则需要引入200bp的无关序列才能重组成环,因此本发明方法的测序成本更低、reads的有效性更高。
[0115]
综上所述,本发明方法在各过程的处理中展现了比10xr2c2、scnaumi-seq 和scnapbar更强大的能力和更高的reads产生效率;比r2c2更高的矫正成功率和更有效的reads产出模式。
[0116]
实施例四:本发明方法的性能效果
[0117]
本实施例以相同cdna样本分别进行了直接ont测序和本发明方法(以 plp环化方式),对亚读长覆盖率对共识序列的碱基质量的影响进行评估。ont 测序的reads显示缺失突变率为1.57%,碱基错配率为3.37%,而本发明方法(以 plp环化方式)的共识序列具有较低的缺失突变和碱基错配,在大于10条亚读长覆盖率下达到0.84%的缺失突变和1.8%的碱基错配。碱基质量(bq)值是衡量测序质量的重要指标,质量值越高代表碱基被测错的概率越小,计算公式为 q=-10lgp(q表示质量值,p表示被测错的概率)。值得注意的是,随着亚读长数量的增加,本发明方法(以plp方式环化)的缺失突变率和碱基错配率降低,且集中分布在相应小提琴图的底部(宽度最大)并颜色较浅(对应的碱基质量值大于20),这表明本发明方法(以plp环化方式)的缺失突变率和碱基错配率均集中在较低的数值且碱基被测错的概率低。另外,测得的单个共识序列的准确性与其基础原始reads的平均碱基质量(bq)得分高度相关,这进一步表明本发明方法能够得到高准确性的共识序列(如图3b-3c所示)。
[0118]
除此之外,本实施例还比较了本发明方法(以plp环化方式)和ngs(第二代测序,next-generation sequencing)条形码之间的一致性。分别计算了本发明方法(以plp环化方式)的共识序列中能够匹配的条形码和ngs测到的条形码之间的最小编辑距离(levenshtein distance),即:以比对同一基因为前提,来自本发明方法(以plp方式环化)的reads与具有相同条形码的ngs相比,可容许的错误碱基数(错误包括缺失、插入、替换)。
为了研究编辑距离对条形码拆解的影响,计算了来自本发明方法(以plp方式环化)reads和具有相同条形码的ngs,与同一基因比对的基因的百分比。具体地,当条形码完全匹配时,即编辑距离=0,大约80%的基因被正确比对;当编辑距离大于3时,正确匹配基因的百分比显著低于完全匹配(“编辑距离=0”对应的百分比)(图3d)。因此,选用编辑距离小于3作为可靠条件,本发明方法则可产生约为65%的正确匹配条形码的reads(图3e),约为34.49%的正确匹配umi的reads(图3f)。
[0119]
实施例五:用本发明方法(以plp方式环化)对小鼠脑四个区域进行空间层面的分析研究
[0120]
在本实施例中,本发明方法应用于10
×
genomic空间转录组平台,对小鼠脑四个区域进行空间层面的分析研究(根据实际需求,还可以将本发明提供的方法应用于单细胞测序)。本发明方法(以plp方式环化)促进了空间特异的异构体(isoforms)的鉴定,即应用仅保留超过3条reads的未注释异构体的严格过滤条件(即新的异构体只有被测到3次,才会保留),总共检测到15,318个唯一的异构体,包括已知基因中的609个新异构体和基因间区域中的66个新异构体。
[0121]
实验人员还鉴定了226个空间特异性的异构体,例如rps24基因的两种异构体(rps24-205(ensmust00000223999)和rps24-202 (ensmust00000169826)),其选择性剪接类型为外显子跳跃(skipping exons, se,图4a、4b)。在ont测序和基于本发明方法测得的reads中鉴定了上述两种转录本,本发明方法的缺失突变率(p=2e-16)和碱基错配率(p=2e-16)显著较低(如图4a、4c所示,其中黑点在图4a中表示错配)。在前脑冠状切片(wta) 中,转录本ensmust00000223999在脑室下区(svz)中的表达最高,而转录本 ensmust00000169826(带有跳过的外显子)在白质(wm)中的表达最高(图4d)。
[0122]
此外,实验人员发现rtn1在小脑冠状(wtd)切片的颗粒细胞(gc)、 bergmann胶质细胞/浦肯野细胞层(bg/pc)和分子层(ml)中表现出高表达(图 4e、4g)。而实验人员发现rtn1基因有一对空间特异性亚型开关,在小脑区域之间的短异构体和长异构体具有选择性转录起始位点(tss,图4f),且所述长异构体和所述短异构体具有偏好性,具体表现为:所述长异构体(rtn1-202)在分子层(ml)区域表达,所述短异构体(rtn1-201,rtn1-203)在颗粒细胞(gc) 中表达(图4e)。使用为短异构体和长异构体设计的单独探针的单分子rna荧光原位杂交(smrna-fish)进一步验证了上述模式,发现短异构体(红色荧光) 在gc中共定位,而长异构体(黄色荧光)在ml中共定位(图4h)。这些结果表明,本发明方法(提高了cdna序列的精度)有助于识别空间特异性异构体。
[0123]
接下来,实验人员从长读长reads中提取了大量的剪接连接点(splicejunction)并探索了未注释剪接连接点的大小。几乎所有的内含子被认为包含两个高度保守的双核苷酸;而剪接连接点是指在切断和重接位点处的两旁的基序(motif),在内含子左侧(5’端)的连接点称为供体(donor),在内含子右侧(3’端)的连接点称为受体(acceptor)。在切断内含子序列的5’端之后,供体剪接位点恰好有gt;在切断内含子序列的3’端之前,受体剪接位点恰好有ag(即 gt-ag规范剪接基序(canonical splice site motif));此外,还可以观察到gc-ag、 at-ac、gg-ag、gt-tg、gt-cg、ct-ag等非规范剪接基序。当以需要至少2条reads的支持且仅使用gt-ag、gc-ag和at-ac的经典二核苷酸为过滤条件时,总共检测到53,305个注释(90.45%)和5,631个新剪接点(9.55%)(图 5a、5b)。为了评估这些新的剪接点是否仅仅是
ont测序产生的噪声,实验人员下载了公共表达序列标签(est)/mrna数据集,找到了1,098(19.5%)个新的且验证的剪接点(“验证”),表明这些经过验证的连接点不是因为ont测序错误导致的。基于新的剪接点的供体位点和受体位点是否在ensembl v93中被注释分成了四类:745个(13.23%)连接点供体位点和受体位点都被注释,但没有注释剪接模式;3,300个(58.6%)剪接事件的供体位点和受体位点都具有剪接模式;794 个(14.1%)和792个(14.06%)剪接位点包括未注释的剪接供体或受体(图5a)。
[0124]
为了进一步表征5,631个观察到的新剪接点,实验人员研究了它们的剪接位点概率分数(splice site probability scores)、保守性分数(conservation scores) 和剪接位点基序(motif)的程度。首先,实验人员观察到“注释”、“验证”的混乱程度较低,而新的且未验证的(“新(novel)”)混乱程度高(图5b)。值得注意的是,当比较ont测序和本发明方法之间的剪接位点概率分数时,无论是在受体(p=4.2
×
10-8
,t检验)还是供体(p=4.2
×
10-8
,t检验)中,本发明方法识别的新连接点的剪接位点概率分数高于ont测序(如图5c所示,纵坐标表示剪接位点的最大熵)。接下来,揭示了剪接位点的保守分数(本实施例使用phastcons来进行序列保守性打分):“注释”位点高度保守,“验证”、“新 (novel)”也具有相似的模式,且与随机序列(mm10)不同(图5d),这表明鉴定的新剪接点在脊椎动物中是高度保守的。然后,实验人员还发现注释、验证以及新(novel)剪接点在受体和供体中都具有高度的编码潜力,并且编码能力范围从高到低(图5e)。实验人员发现,与注释位点(99.26%)相比,“验证
”ꢀ
(94.83%)具有比例接近的规范剪接基序gt-ag,而“新(novel)”有一定差异(图5f)。实验人员在ttr基因中,揭示了未注释的外显子跳跃模式,新异构体18:20666587-20673630表现出比ttr基因更窄的表达模式,但与已注释的异构体ensmust00000075312共定位(图5g、5h),这表明上述两个异构体在相同的空间区域中表达。总之,本发明提供的方法能够识别许多具有高剪接分数和高保守性的新剪接点。
[0125]
为了研究小鼠脑中rna编辑的空间分布,实验人员分别从短读(short reads,也可以理解为“二代测序”中的illumina技术)和长读(long reads,也可以理解为“三代测序”中的ont技术)中分别鉴定了13,397和8,533个rna编辑位点(如图6a所示)。通过rediportal[参考文献14]注释发现,这些位点大多数位于alu区域,并且倾向于a-to-i(adenosine-to-inosine,腺嘌呤核苷转为次黄嘌呤核苷)的编辑模式。对于上述两种技术(即,长读长的ont技术与短读长的illumina技术)均找到的rna编辑位点,其rna编辑水平具有高度相关性 (pearson相关系数》0.96)。值得注意的是,与ont测序和illumina之间的差异相比,实验人员发现本发明方法与illumina的rna编辑水平的差异显著较低 (p=0.016,wilcox检验,如图6b所示)。例如,本发明方法(以plp环化方式)准确地识别了pitpna的chr11:75599645和ttc41的chr10:86731780的poly-t 区域中的rna编辑,而ont 1d无法准确识别(与illumina差异明显)(图6c),这表明本发明方法提高了rna编辑定量的准确性。接下来,实验人员在前脑冠状切片中确定了空间特异性rna编辑位点(图6d)。例如,ttc41的chr10:86731780 在皮质层(ctx,cortex)中具有特异性(图6e、6f),而新异构体的16:51669711 仅出现在白质(wm,white matter)区域(图6e、6g)。值得注意的是,vmp1 的11:86630507分布在除皮质(ctx,cortex)和下丘脑(ht,hypothalamus) 之外的大多数区域(图6e、6h)。总之,利用本发明提供的方法,实验人员确定了空间特异的rna编辑位点,这表明本发明方法有助于rna编辑位点的定量。
[0126]
综上所述,本实施例通过将本发明方法应用至空间转录组测序,分析了小鼠脑四个区域的异构体和rna编辑全景,并确定了以空间特异的方式选择性剪接异构体或rna编辑的多个基因。
[0127]
实施例六:
[0128]
基于上述三代测序方法,本发明还提供了一种三代测序系统100,下面结合具体附图和实施例进行说明。
[0129]
图8为本发明的一种三代测序系统的功能模块图,具体地,本实施例的所述三代测序系统100包括:
[0130]
序列信息接收模块:用于接收待测核酸序列的原始串联重复序列信息,所述待测核酸序列的原始串联重复序列信息是通过第三代测序得到,所述第三代测序包括纳米孔测序和/或单分子荧光测序;还可以用于接收所述待测核酸序列的第二代测序信息,所述第二代测序包括离子半导体测序、焦磷酸测序、连接测序、合成测序、组合探针锚点合成测序和可逆性末端终止测序中的一种或多种。其中,所述待测核酸序列包括核酸内嵌序列、标识序列以及分别位于所述待测核酸序列两端的固定序列,所述原始串联重复序列是以环化的所述待测核酸序列为模板,滚环扩增得到。
[0131]
第一匹配模块:用于基于所述固定序列,对所述原始串联重复序列进行第一轮标识匹配,提取标识区域第一序列。
[0132]
第二匹配模块:用于基于所述固定序列和所述标识序列,对所述原始串联重复序列进行第二轮标识匹配,从而对所述标识区域第一序列进行校正,得到标识区域第二序列。需要说明的是,所述第一匹配模块和所述第二匹配模块可以整合为一个匹配模块。
[0133]
拆解模块:用于基于所述标识区域第二序列,对所述原始串联重复序列进行拆解,得到一个或多个亚读长,所述亚读长包括所述核酸内嵌序列的中间产物和所述标识区域第二序列。
[0134]
比对模块:用于对所述亚读长进行多序列比对,提取核酸第一共识序列,所述核酸第一共识序列即视为所述核酸内嵌序列的测序结果;还可以用于在所述第二轮标识匹配前,基于所述标识区域第一序列,对所述原始串联重复序列进行比对,提取核酸第二共识序列。需要说明的是,所述拆解模块和所述比对模块可以整合为一个分析模块。
[0135]
在一具体实施例中,所述序列信息接收模块将接收到的所述待测核酸序列的原始串联重复序列信息,传输给所述第一匹配模块进行所述第一轮标识匹配;所述第一匹配模块将提取的标识区域第一序列信息传输给所述第二匹配模块;所述序列信息接收模块将接收到的所述待测核酸序列的第二代测序信息传输给所述第二匹配模块;所述第二匹配模块对所述原始串联重复序列进行第二轮标识匹配,对所述标识区域第一序列进行校正,并将得到的标识区域第二序列信息传输至所述拆解模块;所述拆解模块基于所述标识区域第二序列,对所述原始串联重复序列进行拆解,得到一个或多个亚读长,并将所述亚读长的信息传输至比对模块;比对模块对所述亚读长进行多序列比对,提取核酸第一共识序列,所述核酸第一共识序列即视为所述核酸内嵌序列的测序结果。此外,所述第一匹配模块还可以将提取的标识区域第一序列信息传输给所述比对模块;所述比对模块在所述第二轮标识匹配前,基于所述标识区域第一序列,对所述原始串联重复序列进行比对,提取核酸第二共识序列。
[0136]
上面结合附图对本发明的实施例进行了描述,但是本发明并不局限于上述的具体实施方式,上述的具体实施方式仅仅是示意性的,而不是限制性的,本领域的普通技术人员在本发明的启示下,在不脱离本发明宗旨和权利要求所保护的范围情况下,还可做出很多形式,这些均属于本发明的保护之内。
[0137]
参考文献1.sharond,tilgnerh,grubertf,snyderm:asingle-molecule long-read survey ofthe humantranscriptome.natbiotechnol2013,31:1009-1014.2.lebrigandk,magnonev,barbryp,waldmannr:high throughputerrorcorrected nanopore single celltranscriptome sequencing.natcommun 2020,11:4025.3.rangfj,kloostermanwp,de ridderj:from squiggle tobasepair:computational approaches for improvingnanopore sequencingreadaccuracy.genomebiol2018,19:90.4.karst sm,ziels rm,kirkegaardrh,sorensenea,mcdonaldd,zhu q,knightr, albertsenm:high-accuracylong-readamplicon sequences usingunique molecular identifiers withnanopore orpacbio sequencing.natmethods 2021,18:165-169.5.wickrr,juddlm,holtke:performance ofneuralnetworkbasecalling tools for oxfordnanopore sequencing.genomebiol2019,20:129.6.voldenr,palmert,byrnea,cole c,schmitz rj,greenre,vollmers c:improving nanoporereadaccuracywiththe r2c2 methodenables the sequencing ofhighly multiplexedfull-length single-cell cdna.procnatlacadsci usa 2018, 115:9726-9731.7.voldenr,vollmers c:highlymultiplexed single-cellfull-length cdnasequencing of human immune cells with 10x genomics andr2c2.biorxiv2020.8.wilsonbd,eisensteinm,sohht:high-fidelitynanopore sequencing ofultra-short dnatargets.analchem 2019,91:6783-6789.9.phospho-rnasequencingwith circaid-p-seq.biorxiv2020.10.marcozzia,jagerm,elferinkm,straverr,van ginkel jh,peltenburgb,chenl-t, renkens i,vankuikj,terhaardc,etal:accurate detection ofcirculating tumordna usingnanopore consensus sequencing.biorxiv2020.11.wang q,s,v,gehring n,altm
ü
ller j,dieterich c:single cell transcriptome sequencing on the nanopore platform with scnapbar rna 2021.12.wright es:using decipher v2.0 to analyze big biological sequence data in r.the r journal 2016,8:352
‑‑
359.13.li c,chng kr,boey ej,ngah,wilma,nagarajan n:inc-seq:accurate single molecule reads using nanopore sequencing.gigascience 2016,5:34.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。