1.本发明涉及精细化工合成技术领域,具体涉及一种甲酚曲唑三硅氧烷的合成新工艺。
背景技术:
2.众所周知,当皮肤接受紫外线过度暴晒后,会永久性损伤皮肤细胞。其中,波长在280-320纳米波段的紫外线(又称uv-b)可加速色素产生,使皮肤变黑;波长在320-400波段的紫外线(又称uv-a)可以穿透至皮肤的真皮层,造成皮肤弹性的损失和加速皱纹的产生,导致皮肤老化;更有异常情形时,则会变成色素性的皮肤癌等。
3.防晒化妆品所使用的防晒剂分为物理防晒剂和化学防晒剂两种,而甲酚曲唑三硅氧烷就是一种常用的新型化学防晒剂,被广泛应用于各种中高端品牌的防晒霜、防晒乳、护发剂等产品,最大添加量为15%;甲酚曲唑三硅氧烷是一种含有机硅结构的新型防晒剂,具有特别好的配方相容性,其最大吸收波长在303纳米和343纳米,基本覆盖了280-400纳米的整个紫外线波段,可以更全面的减少紫外线对皮肤的损伤;同时,由于其分子结构中含有特殊的共轭结构和分子氢键,因此具有长时间的光稳定性,从而更好地提高复配体系的spf值。
4.目前,关于甲酚曲唑三硅氧烷的合成,报道的文献很少,在专利wo2012/055063和专利wo2012/055064中,使用2-(2-羟基-5-甲基苯基)苯并三唑为原料,使用碳酸钾为傅酸剂,水/苄醇(或二苯醚,四氢化萘等高沸点溶剂)为混合溶剂,在相转移催化剂条件下,和甲基烯丙基氯发生醚化反应合成中间体i,然后中间体i再在高于200℃的条件下发生克莱森重排反应合成关键中间体ii,然后中间体ii在卡斯特催化剂条件下和七甲基三硅氧烷发生硅氢加成,合成目标产物,合成路线如下所示;
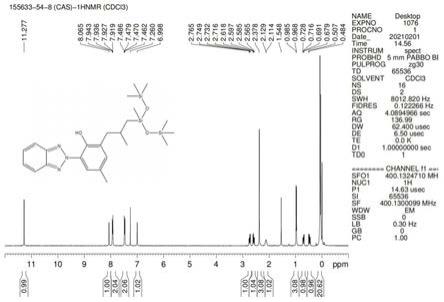
在上述工艺中,第一步采用是两相反应,需要较多的相转移催化剂和过量的碳酸钾等傅酸剂,不仅造成原料的浪费,而且需要很长的反应时间(至少20小时以上),效率很低;第二部采用传统的克莱森重排,温度需要达到200℃以上,同时,克莱森重排在反应引发后是放热反应,一旦温度超过230℃就会进一步重排生成重排杂质a(结构如下所示,杂质a 的hnmr见附图5),在放大生产时,高温不仅增加了工业化的难度和危险性,而且加热反应的同时又是放热反应,很难实现严格控制温度;步骤三采用经典的硅氢加成,由于分子中存在三个n元素,n元素的孤对电子会降低卡斯特催化剂的活性,该步骤原料、反应溶剂和投料过程均需要严格控制无水无氧,无水溶剂不仅价格昂贵而且回收后难以套用,同时反应温度需要严格的控制在65-70℃,温度过高反应催化剂活性会降的更低,转化率也会降低。
5.总而言之,上述工艺每一步小试阶段还可以实现,但是放大到工业化生产,原料过量浪费、生产效率低,工艺参数要求苛刻,危险系数高,同时也很难控制杂质生成,溶剂回收套用困难,造成三废增加。
6.因此,需要提供一种甲酚曲唑三硅氧烷的合成新工艺,以解决上述现有存在的问题。
技术实现要素:
7.有鉴于此,本发明提供一种甲酚曲唑三硅氧烷的合成新工艺,以解决背景技术中存在的问题。
8.为解决上述技术问题,本发明提供一种甲酚曲唑三硅氧烷的合成新工艺,包括以下步骤:步骤一、对甲基苯酚为原料,溶于反应溶剂a,在lewis酸催化条件下,与甲基烯丙基氯回流反应2-3小时,发生付克烷基化反应傅克-烷基化反应合成中间体a;步骤二、步骤一所得中间体a在反应溶剂b中,铂催化剂条件下,与七甲基三硅氧烷发生硅氢加成反应3-5小时,合成中间体b;步骤三、步骤二所得中间体b和邻硝基苯胺发生重氮-耦合反应,经后处理得到中间体c;步骤四、步骤三所得中间体c经还原环化反应,还原直接得到粗品,然后经重结晶得到精品,烘干,包装得终产品。
9.合成路线为:
进一步的,步骤一中,甲基烯丙基氯的使用当量为1.0-1.5;步骤二中,七甲基三硅氧烷使用当量为1.0-2.0;步骤三中,邻硝基苯胺的使用当量为1.0-1.5。
10.进一步的,步骤一中,lewis酸为alcl3、fecl3或zncl2,使用当量为1.0-3.0。
11.进一步的,步骤一中,反应溶剂a为二氯甲烷或二氯乙烷;步骤二中,反应溶剂b为甲苯、乙酸乙酯、四氢呋喃或异丙醇。
12.进一步的,步骤二中,铂催化剂为speier催化剂和karstedt催化剂,其中speier催化剂使用量为50-100ppm,karstedt催化剂使用量为5-10ppm。
13.进一步的,步骤三中,重氮耦合反应重氮盐形成时,使用硫酸/盐酸。
14.进一步的,步骤三中,重氮耦合反应,重氮盐制备时,采用微通道管式反应器,制备好后直接使用。
15.进一步的,步骤四中,还原剂为葡萄糖/锌粉、水合肼/锌粉、水合肼/铁粉、水合肼/保险粉、葡萄糖/氢化或水合肼/氢化。
16.进一步的,步骤四中,催化剂为钯碳(可套用4次以上),反应压力为2-3公斤压力。
17.进一步的,步骤四中,重结晶溶剂为甲醇、乙醇、异丙醇、甲醇水溶液、乙醇水溶液或异丙醇水溶液。
18.本发明的上述技术方案至少包括以下有益效果:1、三步反应都是均相反应,时间短,效率高,成本低;2、没有高温反应,反应安全,同时将高温容易分解得重氮盐使用微通道管式反应器制备,制备好后直接使用,实现0储存,反应可操作性强,转化率高;3、硅氢加成反应中分子内不存在降低催化剂活性的n、p、s等元素,降低了催化剂使用量,催化剂活性稳定,不需要使用严格无水的溶剂,使用常规的普通溶剂即可达到理想效果,降低了原材料成本;4、四步反应几乎均是等当量进行,不造成原材料的浪费,转化率高,精品的总收率
可达80%以上,成本低;5、四步反应均为常规反应,工业化要求低,实现了百公斤级放大生产。
附图说明
19.图1为本发明实施例1合成甲酚曲唑三硅氧烷的核磁共振氢谱;图2为本发明实施例1合成甲酚曲唑三硅氧烷的高效液相色谱;图3为本发明实施例1合成甲酚曲唑三硅氧烷的紫外可见吸收光谱;图4为本发明实施例1中间体a的核磁共振氢谱;图5为杂质a的核磁共振氢谱。
具体实施方式
20.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例的附图1-5,对本发明实施例的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于所描述的本发明的实施例,本领域普通技术人员所获得的所有其他实施例,都属于本发明保护的范围。
21.实施例1步骤一:在250ml三口瓶中,加入对甲基苯酚(21.6g,1.0eq),二氯乙烷66ml,控制温度不超过30℃,分批加入无水三氯化铝(29.2g,1.1eq),加入完毕后搅拌30分钟,后缓慢滴加甲基烯丙基氯(19g,1.05eq),滴加完毕后,升温至65-70℃反应2-3小时,tlc监控反应进程,反应结束后,将反应液降至室温,后分批倒入200 g冰水中淬灭,分出有机相,依次用饱和碳酸氢钠洗涤,饱和盐水洗涤,无水硫酸镁干燥,浓缩至干得棕色油状物,经石油醚溶解,刷硅胶柱纯化得到29g黄色油状物,收率89.5%;1h nmr(400mhz,cdcl3)δppm 11.40(s,1h,oh),7.05(d,1h,arh),6.88(m,1h,arh),6.65(d,1h,arh),4.86(s,1h,c=ch2),4.74(s,1h,c=ch2),3.48(s,2h,ch2),2.38(s,3h,phch3),1.80(s,3h,ch3)。
22.步骤二:在100ml三口瓶中,加入步骤一中的中间体a(16.2g,1.0eq),乙酸乙酯30ml,氮气置换三次,加入1%铂含量的speier催化剂0.16g(以铂计量,为中间体a重量的100ppm),再氮气置换三次,后缓慢滴加七甲基三硅氧烷(22.2g,1.0eq),滴加时略有放热,保持温度不超过55℃,滴加完毕后,搅拌半小时,后升温至回流反应3-5小时,hplc中控反应结束,继续升温蒸出多余得乙酸乙酯,直至内温达到90-100℃,常压已蒸馏不出溶剂时,换机械泵减压继续蒸馏1小时,得到38 g淡黄色油状物,反应几乎为定量反应,粗品直接用于下一步。
23.步骤三:在100ml三口瓶中,加入邻硝基苯胺(6.9g,1.0eq),水10ml,盐酸(16ml,3.0eq),搅拌溶清后,降温至0℃以下,后滴加30%的亚硝酸钠溶液(11.5g,1.0eq),控制温度最高不超过5℃,滴加完毕后,低温搅拌半小时,溶清后备用;在另一个250ml三口瓶中,加入中间体b(19.2g,1.0eq),乙醇30ml,水15ml,搅拌溶解后,控制温度不超过25℃的条件下加入氢氧化钠(2.2g,1.1eq),搅拌溶清后,降温至0℃,然后缓慢滴加备用的重氮盐溶液,最高温度不得高于5℃,滴加完毕后,低温搅拌半小时,后在5℃以下缓慢滴加10%液碱,控制体系ph值维持在8-9,hplc监控反应结束后,缓慢升至室温继续搅拌1-2小时,有大量红色固体析出,抽滤晾干得产品23.5g,收率88.2%。
24.步骤四:在100ml三口瓶中,加入中间体c(10.65g,1.0eq),乙醇50ml,加入30%液碱(2.5g,1.0eq),缓慢升温至65-70℃溶清,后分批加入工业葡萄糖(4.5g,1.25eq),反应液颜色由红色逐渐变为淡黄色,tlc监控反应完成后,5%稀盐酸调ph至5-6,析出黄色沉淀物,过滤得湿品15g,直接溶解于30ml乙醇中,加入5%湿钯碳0.15g,氢气球置换三次,后45-55℃搅拌2-3小时,hplc监控反应结束,滤出钯碳,直接降温结晶,过滤烘干得到黄色终产品9.5g,收率93.2%;1h nmr(400mhz,cdcl3)δppm 11.27(s,1h,oh),8.07(s,1h,arh),7.92-7.94(m,2h,arh),7.46-7.49(m,2h,arh),7.00(s,1h,arh),2.72-2.77(m,1h,ch2),2.56-2.62(m,1h,ch2),2.38(s,3h,phch3),2.11-2.13(d,1h,ch),0.97-0.99(d,3h,ch3),0.68-0.73(m,1h,ch2),0.46-0.51(m,1h,ch2),0.03-0.18(m,21h,ch3);熔点:49.1-49.9℃;hplc保留时间9.09min,纯度98%以上。
25.实施例2步骤一:在2000ml三口瓶中,加入对甲基苯酚(216g,1.0eq),二氯乙烷500 ml,控制温度不超过30℃,分批加入无水三氯化铝(290 g,1.1 eq),加入完毕后搅拌30分钟,后缓慢滴加甲基烯丙基氯(190 g,1.05eq),滴加完毕后,升温至65-70℃反应2-3小时,tlc监控反应进程,反应结束后,将反应液降至室温,后缓慢倒入1500 g水中淬灭,淬灭过程温度不得超过45℃,而后静置分层,分出有机相,用150g水洗一次,无水硫酸钠干燥,有机相浓缩至干得棕色油状物,然后将该油状物用高真空油泵减压蒸馏得到302 g淡黄色油状物,收率93.2%,kf水分含量0.03%。
26.步骤二:在1000 ml三口瓶中,加入步骤一中的中间体a(162g,1.0eq),甲苯200 ml,氮气置换三次,加入8 mg的karstedt催化剂(2%铂含量,以铂计量,为中间体a重量的5ppm),再氮气置换三次,后缓慢滴加七甲基三硅氧烷(220 g,1.0eq),滴加时略有放热,保持温度不超过60℃,滴加完毕后,搅拌半小时,后升温至80℃反应3-5小时,hplc中控反应结束,减压蒸馏蒸出甲苯,直至内温达到120℃不出溶剂时,换机械泵减压继续蒸馏1小时,蒸馏回收的甲苯可测水分后直接套用,得到386 g淡黄色油状物,反应几乎为定量反应,粗品直接用于下一步。
27.步骤三:在1000ml三口瓶中,加入邻硝基苯胺(138g,1.0eq),水200ml,盐酸(320 ml,3.0eq),搅拌溶清后,降温至0℃以下,后滴加30%的亚硝酸钠溶液(230g,1.0eq),控制温度最高不超过5℃,滴加完毕后,低温搅拌半小时,溶清后备用;在另一个2000ml三口瓶中,加入中间体b(384g,1.0eq),乙醇500ml,水300ml,搅拌溶解后,控制温度不超过25℃的条件下加入氢氧化钠(44g,1.1eq),搅拌溶清后,降温至0℃,然后缓慢滴加备用的重氮盐溶液,最高温度不得5℃,滴加完毕后,低温搅拌半小时,后在5℃以下缓慢滴加10%液碱,维持反应体系ph在8-9之间,反应完毕后,缓慢升至室温继续搅拌1-2小时,有大量红色固体析出,抽滤晾干得产品482g,收率90.4%。
28.步骤四:在1000 ml三口瓶中,加入中间体c(212g,1.0eq),乙醇500 ml,加入30%液碱(50 g,1.0eq),缓慢升温至65-70℃溶清,后分批滴加80%水合肼(25g,1.0eq),反应液颜色由红色逐渐变为淡黄色,tlc监控反应完成后,5%稀盐酸调ph至5-6,析出黄色沉淀物,过滤得湿品300g,直接溶解于600 ml乙醇中,加入5%湿钯碳3g,转入加氢釜,氢气置换三次,后在氢气压力为2公斤压力下,45-55℃搅拌2-3小时,hplc监控反应结束,滤出钯碳,直接降温结晶,过滤烘干得到黄色终产品185g,收率93%;熔点:49.0-49.5℃;纯度98以上。
29.实施例3本实施例中,步骤一、二、三的合成步骤与实施例2相同。
30.本实施例与实施例2的区别在于步骤四采用的钯碳为套用回收的,本实施例的目的在于探索钯碳的可套用次数;每次投料212 g中间体c的基础上,套用钯碳可得表1数据表1由表1可知,钯碳套用次数4次,产品收率和品质上无本质差别。
31.实施例4步骤一:在1500l高位反应釜中,加入对甲基苯酚216公斤,二氯乙烷600公斤,控制温度不超过30℃,分批加入无水三氯化铝285公斤,加入完毕后搅拌30分钟,后缓慢滴加甲基烯丙基氯190公斤,滴加完毕后,升温至65-70℃反应2-3小时,tlc监控反应进程,反应结束后,将反应液降至室温;在3000l低位反应釜中放入1500公斤水,后缓慢流入高位反应液淬灭,淬灭过程温度不得超过55℃,而后静置分层,分出有机相,用150公斤水洗一次,有机相常压浓缩至干得棕色油状物,然后将该油状物用高真空罗茨泵,减压蒸馏得到20公斤前馏分(kf水分含量0.15%)和280公斤淡黄色油状产品(kf水分含量0.03%),收率92.6%。
32.步骤二:在1000 l反应釜中,加入步骤一中的中间体a 140公斤,甲苯150公斤,氮气置换三次,加入70 g的卡斯特催化剂(2%铂含量,以铂计量,为中间体a重量的5ppm),再氮气置换三次,后缓慢滴加七甲基三硅氧烷190公斤,滴加时略有放热,保持温度不超过60℃,滴加完毕后,搅拌半小时,后升温至80℃反应3-5小时,hplc中控反应结束,减压蒸馏蒸出甲苯,直至内温达到120℃不出溶剂时,罗茨泵高真空继续蒸馏至不再出液体为止,蒸馏回收的甲苯可测水分后直接套用,得到325公斤淡黄色油状物中间体b,反应几乎为定量反应,粗品直接用于下一步。
33.步骤三:将70公斤邻硝基苯胺、100公斤水,160公斤浓盐酸,搅拌溶解配成溶液备用;将亚硝酸钠35公斤配置成30%溶液,备用;在1000l反应釜中,抽入中间体b 190公斤,乙醇250公斤,然后缓慢加入150公斤20%液碱,搅拌溶清后,降温至0℃,将邻硝基苯胺溶液和亚硝酸钠溶液经计量泵、管式反应器现制备的重氮盐溶液直接滴加至反应釜中,控制计量泵流速,控制反应釜最高温度不得超过5℃,滴加完毕后,低温搅拌半小时,后在5℃以下缓慢滴加10%液碱,维持反应体系ph在8-9之间,反应完毕后,缓慢升至室温,继续搅拌1-2小时,离心得中间体c红色湿品253公斤,直接用于下一步。
34.步骤四:在1000 l反应釜中,加入中间体c125公斤,乙醇240公斤,30%液碱30公斤,缓慢升温至65-70℃溶清,后分批加入工业葡萄糖50公斤,保温反应4-5小时,反应液颜色由红色逐渐变为淡黄色,tlc监控反应完成后,5%稀盐酸调ph至5-6,析出黄色沉淀物,离心得湿品160公斤,将其直接溶解于250公斤乙醇中,加入5%湿钯碳1公斤,转入加氢釜,氢气置换三次,后在2-3公斤压力下,45-55℃搅拌反应2-3小时,hplc监控反应结束,压滤出钯碳,直接降温结晶,离心得到黄色湿品133公斤,hplc保留时间9.10min,纯度98.2%;湿品用220公斤95乙醇经活性炭脱色,重结晶,干燥包装得到105公斤类白色产品,hplc纯度99.5%以上,精品收率89.3%;熔点:49.8-50.2℃。
35.在本发明中,除非另有明确的规定和限定,例如,可以是固定连接,也可以是可拆卸连接,或成一体;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通或两个元件的相互作用关系,除非另有明确的限定,对于本领域的普通技术人员而言,可以根据具体情况理解上述术语在本发明中的具体含义。
36.以上是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。