1.本发明属于去芳构化技术领域,具体涉及到一种通过对苯并呋喃去芳构化制备双键有机化合物的方法。
背景技术:
2.苯并呋喃作为一种常见的芳杂环化合物广泛存在于自然界中,补骨脂素和异补骨脂素都有着这种结构;然而相较吲哚,苯并呋喃的官能团化的探究相对较少。
3.早期的苯并呋喃去芳构化大都局限于直接使用金属烷基试剂对其进行去芳构化,在断裂c-o键的同时引入金属烷基试剂上的基团。但是用该方法引入的基团会受到烷基金属试剂的影响,由于烷基金属试剂的高活性导致该方法只能引入惰性基团,应用价值不高。
4.此后研究者用ni-nhc复合物在100℃的温度下实现了苯并呋喃去芳构硼化,碳硼键作为金属催化交叉偶联最常见的化学键,也为该化合物展现出良好的应用前景,但是反应本身使用的金属复合物直接应用于工业生产并不便利,而且其相对较高的反应温度在一定程度上限制了反应底物的种类。
5.后来研究者使用苯基二甲基硅氢对苯并呋喃进行了去芳构化引入了苯基二甲基硅,苯基二甲基硅作为官能团可以参与金属交叉偶联反应从而替换为其他基团,但该去芳构化的方法还原原先苯并呋喃2,3位的双键成为单键,应用价值相对减少,当前并没有人研究过在保留双键结构的基础上进行金属催化的苯并呋喃去芳构硅化反应。
技术实现要素:
6.本发明的目的是提供一种通过对苯并呋喃去芳构化制备双键有机化合物的方法,可以对苯并呋喃进行去芳构化,同时引入易官能团化的硅基和硼基,反应条件温和,制备工艺简短高效,有效解决了苯并呋喃难去芳构化及难应用等问题。
7.为达上述目的,本发明提供了一种通过对苯并呋喃去芳构化制备双键有机化合物的方法,可以制备出易官能团化硅烷基烯基苯酚,包括以下步骤:
8.(1)将铁催化剂、配体和碱性化合物于密封且无水无氧的条件下混合均匀,并通过氩气进行抽换气后,再加入苯并呋喃、含硼有机化合物和有机溶剂,搅拌条件下制得反应物;
9.(2)将反应物依次进行淬灭、萃取、干燥、过滤、真空浓缩和纯化,即得。
10.进一步地,碱性化合物为叔丁醇钠,铁催化剂为三氟甲磺酸亚铁,配体为1,2-双(二苯基膦基)苯,含硼有机化合物为三乙基(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)硅烷,有机溶剂为四氢呋喃。
11.进一步地,三氟甲磺酸亚铁、1,2-双(二苯基膦基)苯和叔丁醇钠的摩尔比为0.01~0.02:0.01~0.02:0.4~0.7。
12.进一步地,苯并呋喃、三乙基(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)硅烷和三氟甲磺酸亚铁的摩尔比为0.1~0.2:0.35~0.7:0.01~0.02,苯并呋喃和四氢呋喃摩尔
与体积比为0.1~0.2:0.5mol/l。
13.进一步地,三乙基(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)硅烷通过以下方法制备得到:在氩气环境中,将联硼酸频那醇酯、[ir(cod)ome]2、4,4'-di-叔丁基联吡啶、三乙基硅烷和四氢呋喃混合搅拌均匀,密闭后在70~90℃温度下加热15~18h,冷却至室温,再依次真空浓缩、纯化和短程蒸馏,得三乙基(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)硅烷;其中,联硼酸频那醇酯、[ir(cod)ome]2、4,4'-di-叔丁基联吡啶和三乙基硅烷摩尔比为50~52:0.1~0.3:0.2~0.5:200~210,其中,联硼酸频那醇酯和四氢呋喃摩尔体积比为50~52:42mol/l;其中将残余物经色谱柱色谱纯化,洗脱剂为石油醚/乙酸乙酯(20/1);在58℃、120toor条件下进行短程蒸馏。
[0014]
进一步地,步骤(1)中的搅拌条件包括:温度为95~105℃、转速为480~550rpm以及反应时间为18~24h。
[0015]
进一步地,步骤(2)具体包括以下步骤:采用饱和氯化铵溶液淬灭反应物后,用乙酸乙酯萃取3次,合并有机相后采用无水硫酸镁干燥,再过滤后真空浓缩至恒重,再用石油醚/乙酸乙酯=20/1进行色谱柱层析。
[0016]
本发明提供了一种通过对苯并呋喃去芳构化制备双键有机化合物的方法,可以制备出易官能团化苯并氧杂硼己环醇,包括以下步骤:
[0017]
(1)将铁催化剂、配体和碱性化合物于密封且无水无氧的条件下混合均匀,并通过氩气进行抽换气后,再加入苯并呋喃、含硼有机化合物和有机溶剂,搅拌条件下制得反应物;
[0018]
(2)将反应物依次进行淬灭、萃取、干燥、过滤、真空浓缩和纯化,即得。
[0019]
进一步地,碱性化合物为叔丁醇锂,铁催化剂为三氟甲磺酸亚铁,配体为1,2-双(二苯基膦基)乙烷,含硼有机化合物为4,4,4',4',6,6,6',6'-八甲基-2,2'-联(1,3,2-二氧硼杂环己烷),有机溶剂为四氢呋喃;
[0020]
三氟甲磺酸亚铁、1,2-双(二苯基膦基)乙烷和叔丁醇锂的摩尔比为0.02~0.04:0.02~0.04:1.5~3.0。
[0021]
进一步地,苯并呋喃、4,4,4',4',6,6,6',6'-八甲基-2,2'-联(1,3,2-二氧硼杂环己烷)和三氟甲磺酸亚铁摩尔比为0.1~0.2:0.25~0.5:0.02~0.04,苯并呋喃和四氢呋喃的摩尔体积比为0.1~0.2:2mol/l。
[0022]
进一步地,步骤(1)中的搅拌条件包括:搅拌温度为65~75℃、转速为480~550rpm以及反应时间为25~32h。
[0023]
进一步地,步骤(2)具体包括以下步骤:采用饱和氯化铵溶液淬灭反应物后,用乙酸乙酯萃取3次,合并有机相后用无水硫酸镁干燥,再过滤后真空浓缩至恒重,再用石油醚/乙酸乙酯=10/1进行色谱柱层析,再用二氯甲烷/乙酸乙酯=50/1进行色谱柱层析,制得苯并氧杂硼己环醇。
[0024]
进一步地,本发明中提到的苯并呋喃可替换为其他类型的苯并呋喃,如5-甲基-苯并呋喃等。
[0025]
进一步地,本发明中提到的四氢呋喃在使用时均需用钠进行预干燥处理。
[0026]
综上所述,本发明具有以下优点:
[0027]
1、本发明实现了苯并呋喃一步去芳构化形成硅烷基烯基苯酚和苯并氧杂硼己环
醇,苯并呋喃在自然界有着广阔的分布,该方法可以快速拓展含苯并呋喃结构的天然分子的结构,为苯并呋喃药物化学方面的应用提供了良好的方法。
[0028]
2、本发明的催化剂为三氟甲磺酸亚铁,该催化剂廉价易得,铁是地球含量最丰富的金属,相较于铜、钯等重金属,铁由于无毒的特性,可以应用于药物生产的最后几步,相较其他金属更具有优势。
[0029]
3、本发明使用的配体为1,2-双(二苯基膦基)乙烷以及1,2-双(二苯基膦基)苯,两者均为常见的双磷配体,便于在工业生产中应用;该反应在克级条件下表现出一定的应用前景。
[0030]
4、铁的价态广泛,-2~ 6价均匀分布,由于其复杂的中间体难以检测,铁催化的反应发展较其他金属更为落后且反应种类较少,本发明使用的铁催化去芳构化的方法拓展了铁催化的反应的应用前景。
附图说明
[0031]
图1为本发明的硅烷基烯基苯酚以及苯并氧杂硼己环醇的合成示意图;
[0032]
图2为实施例1所得产物的1h nmr谱图;
[0033]
图3为实施例1所得产物的13c nmr谱图;
[0034]
图4为实施例2所得产物的1h nmr谱图;
[0035]
图5为实施例2所得产物的13c nmr谱图。
具体实施方式
[0036]
以下结合实施例对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0037]
实施例1
[0038]
本实施例提供了一种通过对苯并呋喃去芳构化制备硅烷基烯基苯酚的方法,包括以下步骤:
[0039]
(1)在手套箱中称取0.02mmol三氟甲磺酸亚铁、0.02mmol 1,3-双(二苯基膦)苯和0.8mmol叔丁醇钠放入耐压封管中,得混合原料,取出后连通双排管采用氩气抽换气3次,每次间隔4min,然后更换软硅胶塞后再用氩气抽换气3次,每次间隔4min;
[0040]
(2)将步骤(1)所得混合原料在氩气氛围中,加入0.2mmol苯并呋喃、0.7mmol三乙基(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)硅烷和0.5ml四氢呋喃,在100℃、520rpm条件下反应24h,得反应物;
[0041]
(3)将步骤(2)所得反应物用饱和氯化铵溶液淬灭,然后用乙酸乙酯萃取3次,每次25ml,合并有机相,无水硫酸镁干燥,过滤后真空浓缩至恒重,再用石油醚/乙酸乙酯=20/1进行色谱柱层析,得(反)-2-(2-(三乙基甲硅烷基)乙烯基)苯酚。
[0042]
上述,三乙基(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)硅烷通过以下方法制备得到:在氩气环境中,将联硼酸频那醇酯、[ir(cod)ome]2、4,4'-di-叔丁基联吡啶、三乙基硅烷和四氢呋喃混合搅拌均匀,密闭后在80℃温度下加热16h,冷却至室温,再依次真空浓缩、纯化和短程蒸馏,得三乙基(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)硅烷,其中,联
硼酸频那醇酯、[ir(cod)ome]2、4,4'-di-叔丁基联吡啶和三乙基硅烷摩尔比为51.3:0.2:0.4:206,联硼酸频那醇酯和四氢呋喃摩尔体积比为51.3:42mol/l。
[0043]
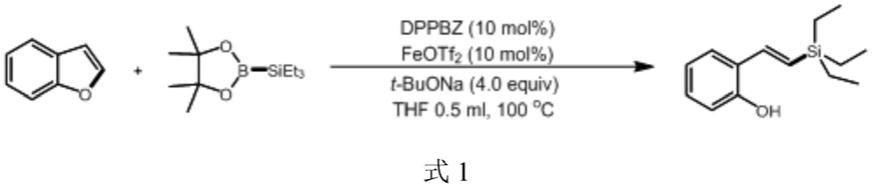
本实施例的反应过程如式1所示:
[0044][0045]
本实施例所得(e)-2-(2-(三乙基甲硅烷基)乙烯基)苯酚产率为78%,将所得产物进行核磁共振测试,其1h、13c核磁共振谱图如图2~3所示。
[0046]
其测试结果如下:1h nmr(400mhz,cdcl3)δ7.43-7.41(d,j=6.8hz,1h),7.19-7.06(m,2h),6.90(t,j=7.5hz,1h),6.78(d,j=8.0hz,1h),6.37(d,j=19.5hz,1h),5.00(s,1h),0.99(t,j=7.9hz,9h),0.66(q,j=7.9hz,6h)。
[0047]
13
c nmr(100mhz,cdcl3)δ152.6,139.0,128.9,128.1,127.0,125.9,120.9,115.9,7.4,3.5。
[0048]
实施例2
[0049]
本实施例提供了一种通过对苯并呋喃去芳构化制备苯并氧杂硼己环醇的方法,包括以下步骤:
[0050]
(1)在手套箱中称取0.04mmol三氟甲磺酸亚铁、0.04mmol 1,2-双(二苯基膦)乙烷和1.5mmol叔丁醇锂放入耐压封管中,得混合原料,取出后连通双排管采用氩气抽换气3次,每次间隔4min,然后更换软硅胶塞后再用氩气抽换气3次,每次间隔4min;
[0051]
(2)将步骤(1)所得混合原料在氩气氛围中,加入0.2mmol苯并呋喃、0.5mmol4,4,4',4',6,6,6',6'-八甲基-2,2'-联(1,3,2-二氧硼杂环己烷)和2ml四氢呋喃,在65℃、450rpm条件下反应24h,得反应物;
[0052]
(3)将步骤(2)所得反应物用饱和氯化铵溶液淬灭,然后用乙酸乙酯萃取3次,每次25ml,合并有机相,无水硫酸镁干燥,过滤后真空浓缩至恒重,用石油醚/乙酸乙酯=10/1进行色谱柱层析,再用二氯甲烷/乙酸乙酯=50/1进行色谱柱层析,得苯并氧杂硼己环醇。
[0053]
本实施例的反应过程如式2所示:
[0054][0055]
本实施例制备得到的2氢-苯并[e][1,2]氧杂硼己环-2-醇产率为77%,将所得产物进行核磁共振测试,其1h、
13
c核磁共振谱图如图4~5所示。
[0056]
其中,1h nmr(400mhz,cdcl3)δ7.78(d,j=11.8hz,1h),7.45-7.32(m,2h),7.14(t,j=7.5,1.2hz,1h),6.21(d,j=11.8hz,1h),4.43(s,1h)。
[0057]
其中,
13
c nmr(100mhz,cdcl3)δ152.27,149.45,129.38,128.69,124.43,122.28,118.42。
[0058]
对比例
[0059]
本对比例提供了一种合成硅烷基烯基苯酚的方法,其合成过程如式3:
[0060][0061]
本对比例所提供的合成硅烷基烯基苯酚的方法与实施例1相比,因为使用了高活性金属有机试剂,反应条件苛刻不易控制,且合成硅烷基烯基苯酚底物受限性大,氧杂烷基烯基苯酚尚无直接合成的方法。
[0062]
虽然对本发明的具体实施方式进行了详细地描述,但不应理解为对本专利的保护范围的限定。在权利要求书所描述的范围内,本领域技术人员不经创造性劳动即可作出的各种修改和变形仍属本专利的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。