一种巴洛沙韦酯晶型d及其制备方法
技术领域
1.本发明是涉及一种巴洛沙韦酯晶型d及其制备方法,属于药物化学技术领域。
背景技术:
2.巴洛沙韦酯(baloxavir marboxil)是日本盐野义制药(shionogi)公司研发的抗流感新药,其商品名为xofluza。xofluza是一款创新的cap依赖型核酸内切酶抑制剂,也是世上少数可以抑制流感病毒增殖的口服新药,它能针对流感病毒复制的关键环节,抑制它从宿主细胞中获得宿主mrna 5’端的cap结构,从而抑制流感病毒自身mrna的转录,该药于2018年2月获日本批准用于成人及儿科患者a型和b型流感的治疗,并于2018年10月获fda批准上市用于治疗12岁及以上不超过48小时的无并发症的急性流感患者。
3.巴洛沙韦酯是前体药物,进入体内水解为活性物质巴洛沙韦,它们的化学结构式如下:
[0004][0005]
根据现有文献的报道,巴洛沙韦酯为多晶型化合物,如:专利wo2018030463中公开了巴洛沙韦酯的form i、form ii和form iii三种晶型,其中的form i晶型是在二甲亚砜与水的混合液中制备得到(详阅其中的实施例10),其中的form ii晶型是先由乙腈(50ml)与水(5ml)的混合液溶清,然后补加95ml水析晶得到(详阅其中的实施例21),其中的form iii晶型是先用40倍乙酸甲酯升温溶解,通过减压浓缩降温析晶得到(详阅其中的实施例22);另外,专利cn201911140898.1中公布了巴洛沙韦酯的form a和form b晶型,其中的form a晶型是先用良溶剂(如:二甲基亚砜、二甲基甲酰胺、甲酸丁酯、丙酮)在常温下溶解,然后在常温下滴加不良溶剂(如:乙醇、水、环己烷、异丙醚)析晶得到(详阅其中的实施例1-4),其中的form b晶型是先用乙腈溶解,然后放置在室温下使乙腈完全挥发获得;由以上现有研究成果可得知,巴洛沙韦酯的晶型对溶剂非常敏感。
[0006]
本专利的发明人在实验中还发现:form ii晶型和form iii晶型均不稳定,在析晶过程中很容易转化为form i晶型;另外,虽然专利cn201911140898.1中公布巴洛沙韦酯的form a和form b晶型在高温、高湿和光照条件下具有很好的稳定性,但是,通过比较我们发现,form a晶型即为专利wo2018030463中公布的form i晶型,而form b的热重分析显示,其有0.5%左右的失重,根据该专利介绍其为无水物,因此此部分失重说明form b是混有溶剂化物的混晶形式。又由于form b是从乙腈体系中挥发析晶得到,而乙腈属于ich规定的二类
溶剂,限度较低,另一方面,混晶也会影响制剂的溶出,因此乙腈溶剂化物形式的form b是不可能作为药剂用的,并且,form b晶型的制备是需要使乙腈溶液在室温下经过3~4天的自然挥发得到,工业生产上肯定实现不了,大量乙腈溶剂挥发到空气中,也不利于环保,而且当量较大时,表层和底层溶剂挥发的程度不一样,更容易出现混晶导致溶剂残留不符合要求的问题。由于上述原因,目前用于口服制剂的原料还是form i晶型(与form a晶型为同种晶型),但现有的form i晶型的晶习成片状,而片状结晶,在烘料的过程中,很容易包裹溶剂而结块团聚,致使烘干后的物料发硬,溶剂残留易超标,往往需要干燥很长时间才能达到溶剂限度,而干燥时间过长,又容易导致降解杂质超标;另外,物料结块后,产品流动性差,需要通过物理粉碎才能达到可压的粒度要求,给口服药物制剂的生产造成了很大困扰。因此,本领域亟需开发在稳定性、溶解性、流动性等方面均优于现有晶型的巴洛沙韦酯新晶型,以满足巴洛沙韦酯口服制剂的工业化生产需求。
技术实现要素:
[0007]
针对现有技术存在的上述问题和需求,本发明的目的是提供一种在稳定性、溶解性、流动性等方面均优于现有晶型的巴洛沙韦酯晶型d,以满足巴洛沙韦酯口服制剂的工业化生产需求。
[0008]
为实现上述发明目的,本发明采用的技术方案如下:
[0009]
本发明所述的巴洛沙韦酯晶型d,为巴洛沙韦酯的无水无溶剂化物晶型,在x射线粉末衍射下,在衍射角2θ为4.5
°
、8.8
°
、10.8
°
、13.2
°
、14.3
°
、14.8
°
和16.2
°
处具有特征衍射峰,测试误差为
±
0.2
°
。
[0010]
进一步说,本发明所述的巴洛沙韦酯晶型d,在x-射线粉末衍射下,在衍射角2θ为4.5
°
、8.8
°
、9.8
°
、10.8
°
、11.7
°
、13.2
°
、14.3
°
、14.8
°
、15.8
°
、16.2
°
、16.5
°
、17.3
°
、17.5
°
、17.8
°
、18.8
°
、20.2
°
、21.6
°
、21.9
°
、22.3
°
、24.2
°
、24.4
°
、26.5
°
、28.0
°
、28.3
°
、29.7
°
和31.5
°
处具有特征衍射峰,测试误差为
±
0.2
°
。
[0011]
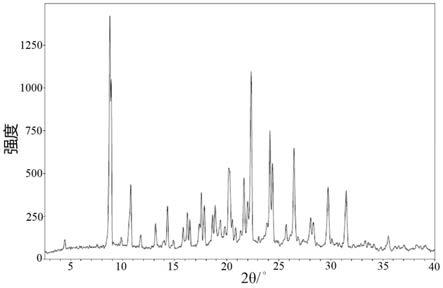
更进一步说,本发明所述的巴洛沙韦酯晶型d的x射线粉末衍射谱图与图4基本一致。
[0012]
进一步说,本发明所述的巴洛沙韦酯晶型d的dsc谱图中,在235℃具有吸热峰。
[0013]
更进一步说,本发明所述的巴洛沙韦酯晶型d的dsc谱图与图5基本一致。
[0014]
更进一步说,本发明所述的巴洛沙韦酯晶型d的热重分析谱图与图6基本一致。
[0015]
一种制备本发明所述的巴洛沙韦酯晶型d的方法,包括如下步骤:
[0016]
a)在20~50℃的溶解温度下,将巴洛沙韦酯原料溶解于由二氯甲烷与酯类溶剂按体积比为1.0:(0.5~2.5)形成的混合溶剂中,得到澄清溶液;
[0017]
b)然后向上述体系中滴加正庚烷,搅拌使析晶;
[0018]
c)过滤收集析出的晶体;
[0019]
d)对步骤c)所得晶体于50~100℃真空干燥6~15小时,即得巴洛沙韦酯晶型d。
[0020]
步骤a)中,所述的溶解温度以20~40℃较优,以25~35℃最优。
[0021]
步骤a)中,所述的巴洛沙韦酯原料的型态不限,可以是无定型或已知的任意一种晶型或它们的混合物。
[0022]
步骤a)中,所述的巴洛沙韦酯原料与所述的混合溶剂的质量体积比为1克:(8~
15)毫升,以1克:(8~12)毫升较优,以1克:10毫升最优。
[0023]
步骤a)中,所述混合溶剂中的二氯甲烷与酯类溶剂的体积比以1.0:(0.8~1.2)较优,以1.0:1.0最优。
[0024]
步骤a)中,所述的酯类溶剂选自苯甲酸甲酯、苯甲酸乙酯、甲酸甲酯、甲酸乙酯、甲酸异丙酯、甲酸丁酯、乙酸丁酯中的至少一种,以选自苯甲酸甲酯、甲酸甲酯、甲酸乙酯中的任意一种为最优。
[0025]
步骤b)中,使用的正庚烷的体积与步骤a)中使用的混合溶剂的体积之比为(1~1.2):1,以1:1最优。
[0026]
步骤d)中,真空干燥的条件以70~90℃下干燥6~10小时较优,以60~80℃下干燥6~8小时最优。
[0027]
与现有技术相比,本发明具有如下显著性有益效果:
[0028]
本发明的研究结果显示:本发明所述的巴洛沙韦酯晶型d是无水无溶剂晶型,晶型的晶习为细长的针状结晶,溶剂容易通过真空干燥除去,干燥后即可得到纯度高达99.5%、杂质残留及溶剂限度均符合原料药质量标准且稳定性、分散性和流动性及可压性均非常好的晶体颗粒,非常适合用作片剂的生产原料;并且,本发明所述的巴洛沙韦酯晶型d在模拟胃、肠液的ph环境中具有优于现有晶型的溶解度,非常有利于口服制剂的吸收利用;总之,本发明所述的巴洛沙韦酯晶型d较现有的已知晶型,具有更适合口服制剂的各项优良性能,相对于现有技术产生了显著进步性和出乎意料的技术效果。
附图说明
[0029]
图1为实施例1中所述晶型c的x-射线粉末衍射图谱(xrpd);
[0030]
图2为实施例1中所述晶型c的差示扫描量热分析谱图(dsc);
[0031]
图3为实施例1中所述晶型c的热重分析数据(tga);
[0032]
图4为实施例1中所述晶型d的x-射线粉末衍射图谱(xrpd);
[0033]
图5为实施例1中所述晶型d的差示扫描量热分析谱图(dsc);
[0034]
图6为实施例1中所述晶型d的热重分析数据(tga);
[0035]
图7为已知晶型i的x-射线粉末衍射图谱(xrpd);
[0036]
图8为实施例4中所述晶型d的稳定性实验的xrpd对比谱图;
[0037]
图9为本技术所述晶型d的晶习照片;
[0038]
图10为已知晶型i的晶习照片。
具体实施方式
[0039]
下面实施例中,除非另有说明,所述的试验方法通常按照常规条件或制造厂商建议的条件实施,所示的原料、试剂均可通过市售购买的方式获得。
[0040]
x-射线粉末衍射的参数如下(xrpd):
[0041]
x-射线粉末衍射仪器:brucker d8 advance x-射线粉末衍射仪;
[0042]
x-射线反射参数:铜靶在室温条件下扫描:
[0043]
电压:40千伏特(kv);
[0044]
电流:40毫安培(ma);
[0045]
扫描模式:连续;
[0046]
扫描范围:2.0~35.0度;
[0047]
步长:0.020
°
;
[0048]
每步测量时间:0.1秒/步;
[0049]
差示扫描量热(dsc)分析方法参数如下:
[0050]
差示扫描量热(dsc)仪器:ta q2000型;
[0051]
温度范围:室温~250℃;
[0052]
扫描速度:10℃/分钟;
[0053]
保护气体:氮气,50毫升/分钟;
[0054]
热重分析(tga)参数如下:
[0055]
热重分析(tga)仪器:tga55型;
[0056]
温度范围:室温~300℃;
[0057]
扫描速度:10℃/分钟;
[0058]
保护气体:氮气,60毫升/分钟。
[0059]
实施例1
[0060]
在室温条件下,将10.0g巴洛沙韦酯溶于50ml二氯甲烷与50ml苯甲酸甲酯形成的混合溶剂中,然后滴加100ml正庚烷,搅拌析晶1小时,过滤,滤饼用正庚烷淋洗,所得到的晶体本技术记为晶型c,其xrpd谱图如图1所示,由图1可见:所述晶型c的x-射线粉末衍射图谱的2θ在4.0
±
0.2
°
、8.0
±
0.2
°
、11.1
±
0.2
°
、11.4
±
0.2
°
、12.0
±
0.2
°
、13.2
±
0.2
°
、13.6
±
0.2
°
、14.1
±
0.2
°
、14.3
±
0.2
°
、16.1
±
0.2
°
、16.3
±
0.2
°
、17.9
±
0.2
°
、20.2
±
0.2
°
、20.8
±
0.2
°
、23.9
±
0.2
°
、24.3
±
0.2
°
、24.5
±
0.2
°
、25.6
±
0.2
°
、28.5
±
0.2
°
和32.6
±
0.2
°
处有特征峰。
[0061]
图2为所述晶型c的差示扫描量热法谱图,由图2可见:所述晶型c在90~100℃和235℃处有吸热峰。
[0062]
图3为所述晶型c的热重分析谱图,由图3可见:所述晶型c在90~100℃有8.0~11.0%的失重,说明所述晶型c为巴洛沙韦酯的苯甲酸甲酯的溶剂化物,其中苯甲酸甲酯含量在8.0~11.0%。
[0063]
将所得到的晶型c置于80℃的真空干燥箱中干燥6小时,可得到9.2g固体,其xrpd谱图如图4所示,即:在衍射角2θ为4.5
°
、8.8
°
、9.8
°
、10.8
°
、11.7
°
、13.2
°
、14.3
°
、14.8
°
、15.8
°
、16.2
°
、16.5
°
、17.3
°
、17.5
°
、17.8
°
、18.8
°
、20.2
°
、21.6
°
、21.9
°
、22.3
°
、24.2
°
、24.4
°
、26.5
°
、28.0
°
、28.3
°
、29.7
°
和31.5
°
处具有特征衍射峰,测试误差为
±
0.2
°
,本技术对干燥后得到的此晶型记为晶型d。
[0064]
图5为所述晶型d的差示扫描量热法谱图,由图5可见:所述晶型d在235℃有吸热峰;图6为所述晶型d的热重分析谱图,由图6可见:所述晶型d为无水无溶剂晶型。
[0065]
实施例2
[0066]
在室温条件下,将10.0g巴洛沙韦酯溶于50ml二氯甲烷与50ml甲酸乙酯形成的混合溶剂中,然后滴加100ml正庚烷,搅拌析晶1小时,过滤,滤饼用正庚烷淋洗,然后于60℃温度下真空干燥8小时,得到9.6g固体,其xrpd谱图与图4基本一致,其差式扫描量热谱图与图5基本一致,其热重分析谱图与图6基本一致,为本技术中所述的晶型d。
[0067]
实施例3
[0068]
将10.0g巴洛沙韦酯、40ml二氯甲烷和50ml甲酸甲酯加入到反应瓶中,升温至35℃溶清,然后滴加100ml正庚烷,搅拌析晶1小时,然后降温至20℃左右,搅拌过滤,滤饼用正庚烷淋洗,然后于80℃温度下真空干燥6小时,得到9.5g固体,其xrpd谱图与图4基本一致,其差式扫描量热谱图与图5基本一致,其热重分析谱图与图6基本一致,为本技术中所述的晶型d。
[0069]
实施例4:稳定性测试
[0070]
根据药物制剂稳定性试验指导原则,对本技术所述的晶型d(由实施例1-3制备得到)和已知晶型i(参照专利wo2018030463中实施例10制备得到,其xrpd谱图如图7所示,与wo2018030463中图3基本一致)进行影响因素实验,包括高温试验、高湿试验和强光照射试验,考察影响其晶型的稳定性条件:
[0071]
高温试验:分别取晶型d和晶型i样品适量,平铺置称量瓶中,在70℃、rh75%的恒温恒湿箱中放置10天后,取上述样品约100mg,采用粉末x-射线粉末衍射(xrpd)测试其晶型情况,结果见表1和图8所示;
[0072]
高湿试验:分别取晶型d和晶型i样品适量,平铺置称量瓶中,在25℃、rh 92.5%的恒温恒湿箱中放置10天后,取上述样品约100mg,采用粉末x-射线粉末衍射(xrpd)测试其晶型情况,结果见表1和图8所示;
[0073]
光照试验:分别取晶型d和晶型i样品适量,平铺至称量瓶中,在可见光4500lux
±
500lux(vis)、紫外光1.7w*h/m2(uv)的恒温恒湿箱(25℃、rh60%
±
5%)条件下放置10天后,取上述样品约100mg,采用粉末x-射线粉末衍射(xrpd)测试其晶型情况,结果见表1和图8所示。
[0074]
表1稳定性试验结果
[0075]
样品晶型晶型d晶型i高温(70℃、rh75%,10天)仍为晶型d仍为晶型i高湿(25℃、rh 92.5%,10天)仍为晶型d仍为晶型i光照(10天)仍为晶型d仍为晶型i
[0076]
结合表1和图8可见,本技术所述的晶型d具有与晶型i相同的稳定性。
[0077]
实施例5:溶解度测试
[0078]
参照日本药典附录配制如下溶液:
[0079]
ph1.2溶液:取氯化钠2.0g,加水适量溶解后,加盐酸7ml,再加水稀释至1000ml,混匀,即得;
[0080]
ph4.0溶液:将0.05mol/l乙酸溶液与0.05mol/l乙酸钠溶液按16.4:3.6比例混合,即得;
[0081]
ph6.8磷酸盐缓冲液:取磷酸二氢钾1.7g和无水磷酸氢二钠1.775g,加水溶解并定容至1000ml,即得。
[0082]
分别取适量晶型d和晶型i样品,并分别用ph1.2溶液、ph4.0溶液、ph6.8磷酸盐缓冲液溶解制成饱和溶液,然后离心,对上清液进行hplc含量分析,计算出相应的溶解度。具体实验结果见表2所示。
[0083]
表2溶解度实验结果
[0084]
样品晶型晶型d晶型iph1.2,37℃22.68μg/ml20.6μg/mlph4.0,37℃24.98μg/ml19.3μg/mlph6.8,37℃23.54μg/ml18.9μg/ml
[0085]
由表2所示结果可见:本技术所述的巴洛沙韦酯晶型d在模拟胃肠液的ph环境中均具有优于现有晶型i的溶解度,非常有利于口服制剂的吸收利用。
[0086]
另外,分别取适量晶型d和晶型i样品,在显微镜下放大100倍观察它们的晶习得知:
[0087]
图9为本技术所述的晶型d的晶习照片,由图9可见:本技术所述晶型d的晶习为细长的针状结晶,颗粒分散性好,说明具有好的流动性和可压性,非常适合用作片剂的原料;
[0088]
图10为已知晶型i的晶习照片,由图10可见:晶型i的晶习为片状,而片状结晶,在烘料的过程中,容易包裹溶剂而结块团聚,致使烘干后的物料发硬,溶剂残留易超标,往往需要干燥很长时间才能达到溶剂限度,而干燥时间过长,容易导致降解杂质超标;另外,物料结块后,产品流动性差,需要通过物理粉碎达到粒度要求,给片剂的生产制备造成了烦扰,不是片剂制备的理想原料。
[0089]
最后需要在此指出的是:以上仅是本发明的部分优选实施例,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容做出的一些非本质的改进和调整均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
