1.本发明属于深度学习领域,主要用于核糖核酸-蛋白质(rna-binding protein,以下简称为rbp)结合点位的识别方法,相较于以往的方法,本方法提出了以注意力机制与卷积神经网络为基础的深度学习方法,该方法有效的挖掘了rbp序列的潜在特征,提升了识别效率,同时降低了方法的参数量,更加轻量化。
背景技术:
2.核糖核酸(rna),主要存在于生物细胞中,在生命体中发挥着调控基因编码的作用,同时也担任着转录过程后的蛋白质合成模板的角色。而一条rna想要完成蛋白质合成任务,脱离不了rbp的帮助。rbp作为翻译过程中重要的媒介物质,是起到重要作用的关键参与者。它们与编码或非编码rna有高度的互动,调节rna的剪切、多腺苷酸化、稳定性、定位和退化。在一系列的相关研究中发现,rbp与癌症、肿瘤等相关疾病有密切的相关性,例如elavl1结合蛋白在肿瘤的增殖、转移血管生成以及耐药性方面就存在着一定关联。因此,研究rbp与rna的结合有助于对以肿瘤为代表的相关生物学内容做出更好的解释,推动相关研究的发展。
3.随着高通量技术的发展,大量的rna序列数据得以产出。对于数据量大,维度高的生物序列数据,一般的生物实验方法对于确定rbp结合位点需要较高的人力物力成本。因此,基于计算的方法成为了对rbp结合位点识别方法中的应用热点。最近,由于深度学习可以捕获高维数据中的潜在特征,在图像,翻译等领域突破性的进展,rbp序列结合位点的分析也逐渐的应用到了深度学习。
4.在深度学习领域中,卷积神经网络(convolutional neural network,以下简称cnn)成为了主要的应用方法。但是,cnn更多是关注于rbp数据局部的特征,并未能关注全局特征。因此,我们此处利用了多头自注意力机制(multi-head self-attention,简称mha)作为全局特征捕捉手段进行补充。mha早期是作为分析序列化数据,如自然语言数据。随着对mha的不断深入研究,其在图像等领域上逐渐取的了良好的成果。
5.本发明专利基于以上方法做了结合,应用于rbp结合位点的识别之中。调研发现,在本发明之前并没有相同技术应用于rbp位点识别领域之中,具有创新性和原创性
技术实现要素:
6.本发明将rbps结合位点数据构建可学习的数据,再利用多个由mha与cnn共同组成的特征提取层捕获潜在特征,生成高维特征向量,并将此类特征向量输入到基于全连接神经网络的分类器中作判别分析,并与实际标签作比较,同时设置交叉熵损失函数指导模型训练。训练完成后,保存模型参数,调用此模型可实现rbps结合位点判别。本发明中的特征提取层与传统的cnn特征提取层相比,在降低该层参数量并保证甚至提升最终分析效果。
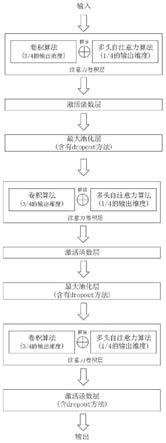
7.本发明针对rbp结合位点识别任务提出了以mha与cnn结合的深度模型(模型结构图见说明书附图1),对从公开数据集库获取的rbp数据集进行数据预处理,首先将长度都统
一为500个碱基,随后利用独热编码(one-hot encoder)的方法将数据中的原有碱基数据进行重新编码。该编码方式可以使计算机有效的识别碱基信息。转化后的数据作为模型训练的原始数据(数据预处理图见说明书附图2)。
8.将原始数据处理完毕后,作为输入数据进入模型中的特征提取层提出其中包含的高位隐藏特征(特征提取部分图见说明书附图3)。该特征提取层由cnn和mha拼接而成,最后在输出时在由一次cnn模块规范输出维度并进一步提取融合特征。整体特征提取曾满足以下公式:
9.h=g
model
(concat[conv(x),mha(x)])
[0010]
其中h为经过特征提取后的高位特征,conv(x)表示卷积特征提取部分,,mha(x)为mha特征提取方法,而x为输入的rbp序列的特征向量。concat表示将两种方法提取出的特征向量进行拼接的方法。
[0011]
其中,在mha中,特征提取满足以下公式:
[0012]
h=g
mutlti-head
(concat[head1(x),head1(x)
…
headn(x)])
[0013]
其中head的个数即表示对输入的rbp序列进行几次自注意力操作,一共有n个头,本发明中n的取值为8。其中每个头中的自注意力机制满足以下公式:
[0014]
其中q=k=v
[0015]
自注意力即是对向量本身的操作,因此q=k=v,同时q,k,v这三个向量是有输入的特征向量x计算得出的,即q=xwq,k=xwk,v=xwv,其中wq、wk和wv为自学习参数,通过训练网络逐渐得出。d为v的维度,在公式中用来降低计算复杂度。
[0016]
在本发明中,输出维度作为单一参数出现,即输出维度由mha和cnn部分拼接而成。本发明中,设计为输出维度的3/4为卷积获得,1/4为mha部分获得。
[0017]
在特征提取层中,激活函数我们采用了tanh函数,添加了最大池化层用来扩大感受野以及利用dropout方法降低过拟合对整体模型的影响。经过上部分特征提取后,会经过扁平层的展平后进入全连接层进行识别分析。
[0018]
本发明中,在最后输出概率上,采用了两层全连接层的设计(全连接层分类器部分图见说明书附图4),并且同样的添加了dropout方法以降低过拟合对整体模型分析效果的影响,激活函数为softmax函数。并且通过引入早停法,在过拟合之前记录下模型训练的最好模型。最终,通过划分出来的测试集验证模型的判断的准确性。
附图说明
[0019]
图1、整体模型结构图。
[0020]
图2、数据预处理图。
[0021]
图3、特征提取模块图。
[0022]
图4、全连接分类器模块图。
具体实施方式
[0023]
以下结合附图和实施例对本发明进行详细说明。
[0024]
步骤1、从相关公开数据集库或相关公开数据网站获取rbp结合位点序列数据
[0025]
步骤2、预处理数据集,利用独热编码对数据进行重新编码,作为模型的输入数据
[0026]
步骤3、将处理好的训练集数据输入到模型当中,利用损失函数和反向传播算法自更新模型参数。
[0027]
步骤4、通过设置的好的早停法,获取最优模型并保存该模型。整体完成后利用测试集进行测试。
[0028]
步骤1的实现过程如下:
[0029]
从公开数据库或公开数据网站下载公共数据集完毕后,将数据集分为正类数据集与负类数据集,正类表示经过生物实验验证为核糖核酸-蛋白质rbp结合位点序列,负类则是从非该位点的序列中随机抽取的序列,保证负类数据集与正类数据集数据量相同。
[0030]
步骤2的实现过程如下:
[0031]
对数据进行预处理,将长度不等的rbp序列同规划成长度为500的序列。针对过长的序列进行剪裁,对长度不足的序列用无意义占位符n在该条序列后补充,并将待操作数据通过独热编码的方式编码成向量化数据。rna序列包含四种碱基,分别为:a(腺嘌呤)、g(鸟嘌呤)、c(胞嘧啶)和u(尿嘧啶),这四种碱基,加上无效占位符n,即有五种字符需要编码,其分别对应编码向量:[1,0,0,0]、[0,1,0,0]、[0,0,1,0]、[0,0,0,1]和[0,0,0,0]。对编码后的数据按4:1的比例进行训练集、测试集划分。
[0032]
步骤3的实现过程如下:
[0033]
根据步骤2获得好的编码数据,输入进网络之中进行训练学习。该网络是由三层特征提取层。每层的特征提取层首先是由多头自注意力机制(multi-head self-attention,以下简称mha)和卷积神经网络(convolutional neural networks,以下简称cnn)组成的特征提取模块,随后是激活函数层,激活函数为tanh,和最大池化层,并配合dropout方法。
[0034]
经过特征提取层后会形成高维特征向量,该向量会经过一层扁平层后进入由两层全连接层组成的分类器中。第一层的全连接层输出维度为512,第二层输出维度2,即最终识别是或不是的概率大小,并依据此概率进行分类判别。为了降低过拟合的影响,同样会配合着dropout方法。
[0035]
步骤4的实现过程如下:
[0036]
整体模型设计好后,便是利用训练数据集配合着交叉熵算法对模型进行训练,完成模型参数自更新。这其中,完成一次正向传播与反向传播为一次epoch。本发明中epoch设置为80。随后利用早停法,当数据在验证集上获得的效果不在上升超过10次epoch后就会停止训练。这其中,验证集是按10%的比例从训练集中划分种出来的。
[0037]
获取到最优模型后,利用测试集数据测试获取到模型效果。其中,训练集和测试集是从原始正负类数据集中划分出来的,训练集与测试集的比例为4:1。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。