1.本技术涉及一种三氮唑基硅烷偶联剂及其制备方法、应用,属于有机硅化合物领域。
背景技术:
2.近年来,聚酰亚胺(pi)、聚苯并噁唑(pbo)等耐热性树脂因其优异的性能而广泛用作液晶显示元件、有机el显示原件、集成电路元件等光学电子部件中的绝缘膜、相位差膜、光学特性膜等固化膜。加热固化后的膜会作为永久膜而存在于器件内,因此,加热后固化膜的性能非常重要。为确保半导体封装的可靠性,固化膜与半导体芯片表面材料的粘合性是非常重要的。
3.随着半导体装置的高集成化、小型化及高速化的要求,近年来的半导体封装的布线向微细化发展,因此正由目前使用的金或铝布线改为电阻更低的铜或铜合金布线。然而,由于聚酰亚胺、聚苯并噁唑等树脂结构本身对基材不具有较强附着力,故耐热性树脂与基材(尤其是铜或铜合金基材)粘合性的提升空间还很大。此外,树脂容易与铜或铜合金发生反应,所以铜或铜合金基材表面易产生变色。
4.在专利文献jp2010152302及专利文献wo2009096050中,公开了包含酰亚胺结构的硅烷偶联剂与具有聚酰亚胺结构的耐热性树脂相容性好,表现出与基材良好的粘合性。专利文献cn111033379a中公开了含酸酐作为官能团的硅烷偶联剂的负型感光性树脂组合物,促使树脂膜与无机材料或金属材料的密合性变得良好。专利文献 cn102375336b中公开了通过在感光性树脂组合物中配合嘌呤衍生物,可获得在铜或铜合金基材上不会引起基材变色的固化膜。而在树脂中直接添加三唑或其衍生物时,由于三唑或其衍生物为小分子化合物,其在树脂高温固化的过程中易挥发,导致抑制铜变色效果不佳。
技术实现要素:
5.本发明的目的是提供一种具有三氮唑基的硅烷偶联剂及其制备方法、应用,该结构的硅烷偶联剂可显著增强聚酰亚胺及聚苯并噁唑等耐热性树脂高温(300℃以上)热固化后与铜或铜合金基材的粘合性,同时可显著改善铜或铜合金基材的变色问题。
6.根据本技术的第一个方面,提供了一种三氮唑基硅烷偶联剂。
7.一种三氮唑基硅烷偶联剂,所述三氮唑基硅烷偶联剂具有如下所示的结构式:
8.g
1-r
1-g29.r1为通式(5)或(6)结构所示的有机基团;
[0010][0011]
通式(6)中,x为通式(5)所示结构;
[0012]
其中,r4为氢原子或碳原子数1~10的烃基;r5为碳原子数4~40的有机基团;
[0013]
g1和g2各自独立地选自通式(1)~(4)所示的结构式中的一种;
[0014][0015]
其中,r2、r2'独立地选自r3o、氢原子、碳原子数1~10的烃基中的至少一种;
[0016]
r3为碳原子数1~10的烃基。
[0017]
可选地,g1和g2为相同的结构。
[0018]
可选地,所述三氮唑基硅烷偶联剂具有通式(i)~(iv)任一所示结构式的单一结构或两种及以上的混合结构:
[0019][0020]
通式(i)~(iv)中,r1、r2、r2'、r3的定义与上述中所述的定义一致。
[0021]
可选地,通式(1)~(4)中,r2、r2'独立地选自r3o、氢原子、碳原子数1~4 的烃基中的至少一种;
[0022]
r3为碳原子数1~4的烃基。
[0023]
可选地,通式(1)~(4)中,r2、r2'独立地选自r3o、氢原子、碳原子数1~4 的烷基中的至少一种;
[0024]
r3为碳原子数1~4的烷基。
[0025]
可选地,通式(1)~(4)中,r2、r2'独立地选自r3o、氢原子、甲基、乙基中的至少一种;
[0026]
r3为甲基或乙基。
[0027]
可选地,通式(1)~(4)中,r2、r2'至少一个为r3o,r3为甲基或乙基。
[0028]
可选地,通式(1)~(4)中,r2、r2'均为r3o,r3为甲基或乙基。
[0029]
可选地,通式(5)中,r4为碳原子数1~3的烃基。
[0030]
可选地,通式(5)中,r4为碳原子数1~3的烷基。
[0031]
可选地,通式(5)中,r4为甲基或乙基。
[0032]
可选地,通式(6)中,r5为碳原子数6~40的含有芳香族环的有机基团。
[0033]
可选地,通式(6)中,r5选自如下结构式中的至少一种:
[0034]
[0035]
其中,r6选自o、c=o、o=s=o、ch
3-c-ch3、cf
3-c-cf3、ch2。
[0036]
根据本技术的第二个方面,提供了一种上述所述的三氮唑基硅烷偶联剂的制备方法。
[0037]
一种三氮唑基硅烷偶联剂的制备方法,包括以下步骤:
[0038]
将通式(7)或(8)所示结构的化合物、通式(9)或(10)所示结构的化合物进行酰胺化反应,或酰胺化反应后再进行亚胺化反应,得到所述的硅烷偶联剂;
[0039][0040]
通式(7)~(10)中,r2、r2'、r3、r4、r5、x的定义与上述中所述的定义一致。
[0041]
可选地,将含有通式(7)或(8)所示结构的化合物、通式(9)或(10)所示结构的化合物的混合物进行酰胺化反应,或酰胺化反应后再进行亚胺化反应,得到所述的硅烷偶联剂。
[0042]
可选地,将通式(7)或(8)所示结构的化合物、通式(9)或(10)所示结构的化合物进行酰胺化反应,或酰胺化反应后再进行亚胺化反应,得到所述的硅烷偶联剂。
[0043]
可选地,包含通式(7)或(8)所示结构的化合物与通式(9)或(10)所示结构的化合物的混合物进行酰胺化反应得到通式(i)或(iii)所示结构的三氮唑基硅烷偶联剂的步骤,以及酰胺化反应后再进行亚胺化反应,得到通式(ii)或(iv)所示结构的三氮唑基硅烷偶联剂的步骤。
[0044]
可选地,通式(7)或(8)所示结构的化合物与通式(9)或(10)所示结构的化合物进行酰胺化反应得到通式(i)或(iii)所示结构的三氮唑基硅烷偶联剂的步骤,以及酰胺化反应后再进行亚胺化反应,得到通式(ii)或(iv)所示结构的三氮唑基硅烷偶联剂的步骤。
[0045]
可选地,通式(7)或(8)所示结构的化合物与通式(9)或(10)所示结构的化合物的摩尔比为2:0.9~1.1。
[0046]
可选地,所述酰胺化反应或亚胺化反应在非质子极性溶剂中进行。
[0047]
可选地,所述非质子极性溶剂选自n-甲基吡咯烷酮、n,n-二甲基甲酰胺、n,n
‑ꢀ
二甲基乙酰胺、二甲基亚砜和γ-丁内酯中的至少一种。
[0048]
可选地,酰胺化反应后再进行亚胺化反应包括:
[0049]
酰胺化反应后在反应液中加入碱和酸酐进行亚胺化反应;
[0050]
所述碱选自吡啶、三乙胺、二异丙基乙基胺中的至少一种;
[0051]
所述酸酐选自乙酸酐、三氟乙酸酐中的至少一种。
[0052]
可选地,所述碱的用量为通式(7)或(8)所示化合物摩尔量的2~10倍,所述酸酐的用量为通式(7)或(8)所示化合物摩尔量的2~10倍。
[0053]
可选地,所述酰胺化反应或亚胺化反应的条件独立地为:
[0054]
反应温度0~100℃,反应时间10~40hr。
[0055]
可选地,所述酰胺化反应或亚胺化反应的条件独立地为:
[0056]
反应温度20~50℃,反应时间15~30hr。
[0057]
可选地,所述酰胺化反应或亚胺化反应的条件独立地为:
[0058]
反应温度20~30℃,反应时间15~25hr。
[0059]
可选地,所述酰胺化反应或亚胺化反应的条件独立地为:
[0060]
反应温度25℃,反应时间20hr。
[0061]
可选地,所述酰胺化反应在25℃下进行。
[0062]
可选地,所述亚胺化反应在25℃下进行。
[0063]
可选地,所述酰胺化反应或亚胺化反应的温度独立地选自0℃、10℃、20℃、25℃、 30℃、35℃、40℃、50℃、60℃、70℃、80℃、90℃、100℃中的任意值或任意两者之间的范围值。
[0064]
可选地,所述酰胺化反应或亚胺化反应的时间独立地选自10h、15h、20h、25h、 30h、35h、40h中的任意值或任意两者之间的范围值。
[0065]
可选地,反应结束后还包括后处理的步骤。
[0066]
可选地,所述后处理包括:对反应液除去溶剂以及其他杂质,蒸馏得到产物。
[0067]
可选地,采用旋转蒸发的方法从反应液中除去溶剂。
[0068]
可选地,利用高温减压蒸馏除去杂质。
[0069]
根据本技术的第三个方面,提供一种树脂组合物。该树脂组合物固化后形成的树脂固化膜可以用作半导体装置的表面保护层、层间绝缘层、再配线层等。
[0070]
一种树脂组合物,包括耐热性树脂和硅烷偶联剂;
[0071]
所述硅烷偶联剂选自上述所述的三氮唑基硅烷偶联剂、上述所述的制备方法制备得到的三氮唑基硅烷偶联剂中的至少一种。
[0072]
可选地,所述硅烷偶联剂的用量为耐热性树脂的质量的0.1~20%。
[0073]
可选地,所述硅烷偶联剂的用量为耐热性树脂的质量的1~10%。
[0074]
可选地,所述耐热性树脂包括聚酰亚胺、聚酰亚胺前体、聚苯并噁唑、聚苯并噁唑前体、聚酰胺、聚酰胺酰亚胺、聚苯并咪唑、聚苯并噻唑树脂中的至少一种。
[0075]
可选地,所述聚酰亚胺前体选自聚酰胺酸或聚酰胺酸酯。
[0076]
可选地,所述聚苯并噁唑前体选自聚羟基酰胺。
[0077]
可选地,所述聚酰亚胺前体的重均分子量为1.8~2.2万。
[0078]
可选地,所述聚苯并噁唑前体的重均分子量为2.1~2.5万。
[0079]
作为一种优选的实施方式,当耐热性树脂为聚酰亚胺前体时,所述树脂组合物的制备方法包括:
[0080]
将含有聚酰亚胺前体、三氮唑基硅烷偶联剂、光引发剂、热交联剂、热阻聚剂的混合物,压滤,得到所述树脂组合物。
[0081]
作为一种优选的实施方式,当耐热性树脂为聚苯并噁唑前体时,所述树脂组合物的制备方法包括:
[0082]
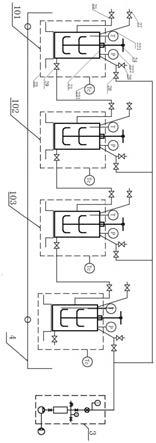
将含有聚苯并噁唑前体、三氮唑基硅烷偶联剂、感光剂的混合物,压滤,得到所述
树脂组合物。
[0083]
本技术中,三氮唑基硅烷偶联剂可以直接添加到树脂或树脂前体中。如对于聚酰亚胺树脂和聚酰亚胺树脂前体,三氮唑基硅烷偶联剂可以直接添加到聚酰亚胺树脂中,得到树脂组合物;三氮唑基硅烷偶联剂也可以直接添加到聚酰亚胺树脂前体中,得到树脂组合物。两种树脂组合物后续会分别经过不同的工序,最终制备固化膜。
[0084]
根据本技术的第四个方面,提供一种三氮唑基硅烷偶联剂作为耐热性树脂改性剂的应用。
[0085]
上述所述的三氮唑基硅烷偶联剂、上述所述的制备方法制备得到的三氮唑基硅烷偶联剂中的至少一种作为耐热性树脂改性剂的应用。
[0086]
本发明所提供的三氮唑基硅烷偶联剂作为耐热性树脂改性剂,用于增强耐热性树脂与铜或铜合金基材的粘合性及改善铜或铜合金基材的变色问题。还可以用于增强耐热性树脂与硅片、玻璃、铜或铜合金以外的其他金属或金属合金等多种基材的粘合性。
[0087]
本技术能产生的有益效果包括:
[0088]
本技术所提供的三氮唑基硅烷偶联剂,作为耐热性树脂改性剂,可显著增强聚酰亚胺及聚苯并噁唑等耐热性树脂高温热固化后与铜或铜合金基材的粘合性,同时可显著改善铜或铜合金基材的变色问题。此外,本技术所提供的三氮唑基硅烷偶联剂的制备方法,简便易行。
附图说明
[0089]
图1为二次布线(rdl)结构示意图。
[0090]
图2为rdl切割方向示意图。
[0091]
图3为二次布线(rdl)中树脂固化膜与rdl剥离程度评价基准。
[0092]
图4是二次布线(rdl)中未添加硅烷偶联剂(图4a)及添加本发明中硅烷偶联剂a-1(图4b)的树脂固化膜与铜基材剥离程度效果图。
具体实施方式
[0093]
下面将对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0094]
本发明提供了一种三氮唑基硅烷偶联剂,所述三氮唑基硅烷偶联剂具有如下所示的结构式:
[0095]g1-r
1-g2[0096]
g1和g2各自独立地选自通式(1)~(4)所示的结构式中的一种;
[0097][0098]
具体地,所述三氮唑基硅烷偶联剂具有通式(i)~(iv)任一所示结构式的单一结构或两种及以上的混合结构:
[0099]
[0100][0101]
式g
1-r
1-g2、(1)~(4)、(i)~(iv)中,r1为通式(5)或(6)结构所示的有机基团,r2、r2'分别独立地选自r3o、氢原子或碳原子数1~10的烃基中的至少一种,r3为碳原子数1~10的烃基;式(5)中,r4为氢原子或碳原子数1~10的烃基;式(6)中,r5为碳原子数4~40的有机基团;x为通式(5)所示结构。
[0102]
具体来说,r2可以为r3o,也可以为氢原子,也可以为碳原子数1~10的烃基,其中,碳原子数1~10的烃基可以为甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、己基、庚基、辛基、壬基、癸基等烷基,环戊基、环己基等环烷基,苯基、甲苯基等芳香基团,苄基、苯乙基、苯基丙基等芳烷基,乙烯基、烯丙基、丁烯基、丙烯基、异丙烯基、苯烯基等不饱和烯基,优选为烷基,更优选为甲基或乙基。r2'可以为r3o,也可以为氢原子,也可以为碳原子数1~10的烃基。r2和r2'相互独立,互不影响。例如,r2和r2'可以同时为r3o,也可以同时为甲基,或者也可以r2为r3o,r2'为甲基。 r3可以为碳原子数1~10的烃基,其中,碳原子数1~10的烃基可以为甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、己基、庚基、辛基、壬基、癸基等烷基,环戊基、环己基等环烷基,苯基、甲苯基等芳香基团,苄基、苯乙基、苯基丙基等芳烷基,乙烯基、烯丙基、丁烯基、丙烯基、异丙烯基、苯烯基等不饱和烯基,优选为烷基,更优选为甲基或乙基。
[0103]
r1为通式(5)或(6)结构所示的有机基团;
[0104][0105]
通式(6)中,x为通式(5)所示结构;
[0106]
其中,r4为氢原子或碳原子数1~10的烃基;r5为碳原子数4~40的有机基团;
[0107]
所述通式(5)中,r4为氢原子或碳原子数1~10的烃基,其中,碳原子数1~10 的烃基可以为甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、己基、庚基、辛基、壬基、癸基等烷基,环戊基、环己基等环烷基,苯基、甲苯基等芳香基团,苄基、苯乙基、苯基丙基等芳烷基,乙烯基、烯丙基、丁烯基、丙烯基、异丙烯基、苯烯基等不饱和烯基,优选为烷基,更优选为甲基或乙基。
[0108]
所述通式(6)中,r5为碳原子数4~40的有机基团,进一步优选为含有芳香族环的碳原子数6~40的有机基团,例如,可以为下述式(11)所示的结构,但并不限定于这些。
[0109][0110]
所述通式(6)中,x为通式(5)所示的结构。由于通式(5)中有取代基r4,根据r4位置的不同,通式(6)的结构有以下三种异构体:
[0111][0112]
所述通式(iii)和(iv)所示的结构中,两侧的硅氧烷基丙基基团可以分别独立地连接在通式(13)所示的不同的位置,即双键两端的5、6、5
′
、6
′
位置和桥环上 1、4、1
′
、4
′
的位置。
[0113][0114]
所述具有通式(i)~(iv)任一所示结构式的单一结构或两种及以上的混合结构,
指的是通式(i)~(iv)所示结构的三氮唑基硅烷偶联剂可以各自单独使用,也可以两种及以上混合使用。当混合使用时,在同一通式或不同通式中以同一符号表示的结构彼此可以相同也可以不同。例如,现有一种三氮唑基硅烷偶联剂是由通式(i)和通式(ii)所示结构的化合物混合而成,则在通式(i)中,r1可以为通式(5)所示的结构,r2为氢原子、r2'为甲基,r3为甲基,在通式(ii)中,r1可以为通式(5)所示的结构,r2为氢原子、r2'为甲基,r3为甲基;r1也可以为通式(6)所示的结构, r2为甲基,r2'为甲氧基,r3为甲基。
[0115]
进一步的,式(1)~(4)、(i)~(iv)中,r2、r2'独立地为r3o、氢原子或碳原子数1~4的烃基,r3为碳原子数1~4的烃基;优选地,r2、r2'独立地为r3o、氢原子或碳原子数1~4的烷基,r3为碳原子数1~4的烷基;更优选地,r2、r2'独立地为 r3o、氢原子、甲基或乙基,r3为甲基或乙基;更优选地,r2、r2'均为r3o,r3为甲基或乙基。此处,r2、r2'、r3及相关术语的含义与前述相同,在此不再赘述。
[0116]
进一步地,通式(5)和(6)中,r4为氢原子,r5为碳原子数4~40的有机基团。更进一步地,通式(5)和(6)中,r4为氢原子,r5为碳原子数6~40的含有芳香族环的有机基团。
[0117]
进一步地,通式g
1-r
1-g2、(i)~(iv)中,r1为通式(5)所示的有机基团。通式(5)中,r4为氢原子或碳原子数1~10的烃基,其具体含义与前面所述一致,在此不再赘述。优选地,r4为氢原子或碳原子数1~3的烃基,进一步优选为氢原子或碳原子数1~3的烷基,更优选为甲基或乙基,最优选为氢原子。例如,r1可以为3,5-二氨基-1,2,4-三氮唑、1-甲基-3,5-二氨基-1,2,4-三氮唑、1-乙基-3,5-二氨基-1,2,4-三氮唑等。
[0118]
在某些具体实施例中,通式(i)~(iv)所示结构的三氮唑基硅烷偶联剂可以是以下结构:r1为通式(5)所示结构,r2和r2'均为甲氧基,r3为甲基,r4为氢原子; r1为通式(5)所示结构,r2和r2'均为乙氧基,r3为乙基,r4为氢原子;r1为通式(5)所示结构,r2和r2'均为戊氧基,r3为戊基,r4为氢原子;r1为通式(5)所示结构,r2和r2'均为辛氧基,r3为辛基,r4为氢原子;r1为通式(5)所示结构,r2为甲基,r2'为甲氧基,r3为甲基,r4为氢原子;r1为通式(5)所示结构,r2为甲基, r2'为乙氧基,r3为乙基,r4为氢原子;r1为通式(5)所示结构,r2和r2'均为甲氧基,r3为甲基,r4为甲基。这些三氮唑基硅烷偶联剂具有很好的提升耐热性树脂和基材的粘合性的作用。其中,r1为通式(5)所示结构,r2和r2'中至少一个为r3o,r3为碳原子数1~10的烷基,r4为氢原子时的硅烷偶联剂的性能更优。
[0119]
本发明还提供了一种通式(i)~(iv)所示的三氮唑基硅烷偶联剂的制备方法,通式(7)或(8)所示结构的化合物与通式(9)或(10)所示结构的化合物进行酰胺化反应得到通式(i)或(iii)所示结构的三氮唑基硅烷偶联剂的步骤,以及酰胺化反应后再进行亚胺化反应,得到通式(ii)或(iv)所示结构的三氮唑基硅烷偶联剂的步骤。
[0120]
其中,通式(7)所示结构的化合物为一种硅烷偶联剂,例如,其可以为3-(三甲氧基硅烷基)丙基丁二酸酐(商品名:x-12-967c,日本信越化学)。
[0121]
通式(8)所示结构的化合物为一种硅烷偶联剂,例如,其可以为5-(三甲氧基硅丙基)-双环[2.2.1]-5-庚烯-2,3-二甲酸酐(又名5-(三甲氧基硅丙基)纳迪克酸酐,简称sanah),其合成方法可以参考日本专利jp2005350655a中的方法进行制备。通式(7)或(8)所示端基为丁二酸酐的硅烷化合物与通式(9)或(10)所示的三氮唑基二胺化合物进行酰胺化反应,得到聚酰胺酸化合物,即通式(i)或(iii)所示结构的三氮唑基硅烷偶联剂。
[0122]
通式(7)或(8)所示端基为丁二酸酐的硅烷化合物与通式(9)或(10)所示的三氮唑
基二胺化合物进行酰胺化反应后再进行亚胺化反应,则得到通式(ii)或(iv) 所示结构的三氮唑基硅烷偶联剂。所述酰胺化反应后再进行亚胺化反应,若亚胺化反应不完全,则得到通式(i)和(ii)所示结构的三氮唑基硅烷偶联剂的混合物,或(iii) 和(iv)所示结构的三氮唑基硅烷偶联剂的混合物。所述亚胺化反应不完全的产物也在本发明的保护范围之内。
[0123]
所述酰胺化反应或亚胺化反应后,还包括对反应液进行后处理得到式(i)或(iii)、式(ii)或(iv)所示的三氮唑基硅烷偶联剂产品的步骤。酰胺化反应后,对反应液直接进行后处理则得到酰胺酸化合物,即式(i)或(iii)所示的硅烷偶联剂;在酰胺化反应后,将得到的反应液继续进行亚胺化反应,待亚胺化反应结束后,对得到的反应液进行后处理,则得到式(ii)或(iv)所示的三氮唑基硅烷偶联剂。所述反应液的后处理包括除去溶剂以及其他杂质,蒸馏得到产物的步骤。可以采用旋转蒸发的方法从反应液中除去溶剂,利用高温减压蒸馏除去杂质,杂质主要成分是未反应的原料以及反应形成的副产物。
[0124][0125]
通式(7)~(10)中,r2、r2'、r3、r4、r5、x的定义与前面所述的定义一致。其中,通式(10)所示的二胺化合物可以通过含有r5基团的二酸酐与通式(9)所示的二胺依次进行酰胺化及亚胺化反应得到。二酸酐可以是任意可以用于酰胺化及亚胺化反应的二酸酐。
[0126]
进一步地,酰胺化反应以及亚胺化反应在非质子极性溶剂中进行,各非质子极性溶剂的效果相当。考虑到成本以及获取方式的便利性,优选地,所述非质子极性溶剂选自n-甲基吡咯烷酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜和γ-丁内酯中的至少一种,优选为n-甲基吡咯烷酮、n,n-二甲基甲酰胺。
[0127]
通式(7)或(8)所示结构的化合物与通式(9)或(10)所示结构的化合物的摩尔比为2:0.9~1.1;优选地,通式(7)或(8)所示结构的化合物与通式(9)或(10) 所示结构的化合物的摩尔比为2:1.
[0128]
酰胺化以及亚胺化反应的温度为25℃,在室温下就可以进行反应,条件温和。
[0129]
进一步地,所述酰胺化反应后再进行亚胺化反应,优选为酰胺化反应后在反应液中加入碱和酸酐进行亚胺化反应;优选地,所述碱为吡啶、三乙胺或二异丙基乙基胺,所述酸酐为乙酸酐或三氟乙酸酐;优选地,所述碱的用量为通式(7)或(8)所示化合物摩尔量的2~10倍,所述酸酐的用量为通式(7)或(8)所示化合物摩尔量的2~10 倍。
[0130]
具体来说,在酰胺化反应后直接在反应液中加入碱和酸酐,将酰胺化反应得到的酰胺酸化合物再进行亚胺化反应,则得到通式(ii)或(iv)所示结构的三氮唑基硅烷偶联剂。所述的碱可以为现有技术中报道的任意可以用于催化亚胺化的碱,例如吡啶、三乙胺或
二异丙基乙基胺等,优选为吡啶,所述碱的用量为通式(7)或(8)所示化合物摩尔量的2倍及以上,例如2~10倍,即可以为2倍、3倍、4倍、5倍、6 倍、7倍、8倍、9倍、10倍。所用的酸酐可以是现有技术中报道的任意可以用于亚胺化的酸酐,例如乙酸酐、三氟乙酸酐等,优选为乙酸酐。所述酸酐的用量为通式(7) 或(8)所示化合物摩尔量的2倍及以上,例如2~10倍,例如可以是2倍、3倍、4 倍、5倍、6倍、7倍、8倍、9倍、10倍。
[0131]
进一步地,本发明的三氮唑基硅烷偶联剂可以作为耐热性树脂改性剂使用,所述三氮唑基硅烷偶联剂的加入可显著增强聚酰亚胺及聚苯并噁唑等耐热性树脂与铜或铜合金基材的粘合性;同时,该三氮唑基硅烷偶联剂还可以显著改善铜或铜合金基材的变色问题。树脂中的某些活性基团例如酯基或羧酸基团易与铜或铜合金基材发生反应,引起基材变色,影响电子器件性能,而三氮唑类基团可以抑制活性基团与基材发生反应,从而改善了铜或铜合金变色的问题,其具体作用机理尚不清楚。本发明中的三氮唑基硅烷偶联剂同时具有三氮唑基团和酰亚胺结构,其与聚酰亚胺及聚苯并噁唑等耐热性树脂相容性好,而且,其中的硅氧烷基团与铜或铜合金基材反应,增强了树脂与基材的粘合性,拉近了硅烷偶联剂分子中三氮唑基团与基材表面的距离,使得三氮唑基团可以聚集在基材表面,同时,硅烷偶联剂为大分子,不易挥发,可以稳定的存在于树脂中,从而更好的抑制铜或铜合金基材变色。本发明中的三氮唑基硅烷偶联剂中的硅氧烷基团的存在对三氮唑基团抑制铜变色有促进作用。值得注意的是,本发明的三氮唑基硅烷偶联剂作为耐热性树脂改性剂,还可以用于增强耐热性树脂与硅片、玻璃、铜或铜合金以外的其他金属或金属合金等多种基材的粘合性。
[0132]
所述耐热性树脂可以为聚酰亚胺树脂、可作为聚酰亚胺前体的聚酰胺酸或聚酰胺酸酯、聚苯并噁唑树脂、可作为聚苯并噁唑前体的聚羟基酰胺、聚酰胺、聚酰胺酰亚胺、聚苯并咪唑、聚苯并噻唑等。
[0133]
硅烷偶联剂在树脂中加入量过低则性能改善效果差,含量过高则会导致树脂组合物的其它性能降低。为了保证高温处理后耐热性树脂与基材的粘合性,硅烷偶联剂的用量为耐热性树脂质量的0.1~20%时较好,例如,用量可以是耐热性树脂质量的0.1%、 0.5%、1%、3%、5%、8%、10%、12%、15%、18%、20%,更优选为1~10%。
[0134]
进一步的,本发明提供了一种树脂组合物,所述树脂组合物包括耐热性树脂和硅烷偶联剂,所述硅烷偶联剂为通式(i)~(iv)所示结构的三氮唑基硅烷偶联剂,所述耐热性树脂定义与前面所述一致。所述树脂组合物固化后形成的树脂固化膜可以用作半导体装置的表面保护层、层间绝缘层、再配线层等,在此不对其进行限定。
[0135]
如无特别说明,本技术的实施例中的原料和催化剂均通过商业途径购买,如无特别说明,测试方法均采用常规方法,仪器设置均采用厂家推荐的设置。
[0136]
实施例中的树脂的评价及硅烷偶联剂添加到树脂中的效果评价按照以下方法进行。
[0137]
(1)分子量测试
[0138]
用凝胶渗透色谱法(标准聚苯乙烯换算)测定树脂的重均分子量(mw)。测定中使用的色谱仪为日本岛津公司的lc-20ad,色谱柱为昭和电工的kf-804,检测器为日本岛津公司的示差rid-20a。
[0139]
(2)粘度测试
[0140]
取0.5ml树脂组合物样品放入旋转粘度计(brookfield dv2t rv)样品池中,选取合适的量程范围,温度控制在25
±
0.1℃,进行粘度测试。
[0141]
(3)树脂固化膜与铜基板的粘合性剥离试验
[0142]
利用匀胶机将树脂组合物样品均匀的涂覆到铜材质的基板上,将其放在120℃的加热台上进行3分钟软烘,得到膜厚为10~20um的树脂膜。利用划格器(byk-gardnera-5125)将树脂膜刻画出10行
×
10列的方格,然后将该膜放置在真空无氧烤箱 (molzk-32d1)中进行热处理:在170℃下热处理30分钟,之后,经过1小时升温至320℃,并在320℃下处理1小时,最终得到固化膜。最后,用胶带(专用透明3m 胶带)参照国家标准gb/t 9286-1998色漆和清漆漆膜的划格试验进行剥离试验,记录下剥离下的格数,作为pct试验前的剥离情况。
[0143]
按照上述同样的方法得到的固化膜,将其放在pct试验箱中进行100小时的pct 老化试验(121℃、2个大气压饱和蒸汽;东莞泓进科技pct-30),pct试验完成后,再采用上述同样的方法利用胶带进行剥离试验,记录下剥离下的格数,作为pct试验后的剥离情况。
[0144]
粘合性剥离实验剥下的个数低于5个时视为“最佳”,低于10个时视为“佳”,低于30个时视为“略佳”,大于等于30个时视为“差”。
[0145]
(4)铜变色试验
[0146]
将树脂组合物均匀的涂覆到铜基板上,然后放在120℃的加热台上进行3分钟软烘,得到膜厚为10~20um的感光性树脂膜,最后将该树脂膜放到显影液中进行溶解。根据以下的基准来评价树脂膜溶解后的铜基板变色情况。
[0147]“最佳”:即使在目视下用200倍的光学显微镜观察时也未确认到铜基板的变色;
[0148]“佳”:在目视下未确认到铜基板的变色,用200倍的光学显微镜观察时稍微确认到铜基板的变色;
[0149]“略佳”:在目视下未确认到铜基板的变色,用200倍的光学显微镜观察时确认到铜基板的变色;
[0150]“差”:在目视下确认铜基板严重变色。
[0151]
(5)多次布线(rdl)中与rdl铜基材的剥离情况
[0152]
将含有本发明硅烷偶联剂的树脂组合物应用在多次布线(rdl)结构中,现对rdl 结构进行说明。如图1所示,1为硅芯片,2为铝焊盘,3为钝化膜,4为含有本发明硅烷偶联剂的树脂组合物形成的层间绝缘膜,5为连接金属膜,6为多次布线(rdl) 金属cu,7为含有本发明硅烷偶联剂的树脂组合物形成的层间绝缘膜,8为凸点下金属层(ubm),9为焊料凸点。
[0153]
多次布线(rdl)结构的制作方法如下:在硅芯片1上制作具有输入输出用途的铝焊盘2、钝化膜3,3上有通孔,进而在3上制作含有本发明硅烷偶联剂的树脂组合物形成的图案化的层间绝缘膜4;利用溅射工艺制作与铝焊盘2相连的金属膜(ticu) 5,进而利用电镀工艺形成金属布线铜6(rdl1),再制作含有本发明硅烷偶联剂的树脂组合物形成的有一定图案的层间绝缘膜7;最后,制作凸点下金属层(under bumpmetallurgy,ubm)8以及焊料凸点9。实际过程中,在层间绝缘膜7上可以选择进一步形成新的金属布线(即rdl2),通过重复进行上述工序,可以形成2层以上的rdl,从而制成了含有本发明硅烷偶联剂的树脂组合物形成的层间绝缘膜分隔开的多层布线结构。多层布线结构的层数没有上限,大多数情况下是在10层以下。rdl制作完成后,为了检验层间绝缘膜7(本发明中也称之为“树脂固化膜”)与
rdl铜基材的密合性能,沿着焊料凸点9的中心线进行切割(如图2所示),并用扫描电镜sem (kyky-1000g型,真空镀金后进行观察,电镜加速电压为10kv)观察结合界面的剥离情况。根据图3所示的评价基准来评价树脂固化膜与rdl之间的剥离程度。如图3 所示,将树脂固化膜与rdl之间的剥离程度分为严重剥离、中度剥离、轻微剥离、无剥离四个等级,剥离越严重则密合性越差。
[0154]
实施例中,硅烷偶联剂的核磁测试采用仪器bruker 400核磁共振仪测试(tms为内标,dmso为溶剂)。
[0155]
实施例中,硅烷偶联剂sanah为5-(三甲氧基硅丙基)-双环[2.2.1]-5-庚烯-2,3
‑ꢀ
二甲酸酐,又名5-(三甲氧基硅丙基)纳迪克酸酐,参考日本专利jp2005350655a中的方法进行制备:以氯铂酸为催化剂,5-烯丙基纳迪克酸酐(anah,日本丸善石油化学株式会社)和三甲氧基硅烷为原料,通过硅氢加成反应来制备。
[0156]
实施例1
[0157]
硅烷偶联剂a-1的合成:
[0158]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,3,5-二氨基-1,2,4-三氮唑9.91g(0.1mol,阿拉丁化学试剂),开启搅拌,待3,5
‑ꢀ
二氨基-1,2,4-三氮唑充分溶解后,缓慢加入3-(三甲氧基硅烷基)丙基丁二酸酐52.47 g(0.2mol,x-12-967c,信越化学),待加料完成后25℃下继续反应20hr,反应完成后进行减压蒸馏纯化得到硅烷偶联剂a-1。
[0159][0160]
所得硅烷偶联剂a-1结构式如(a-1)所示。
[0161]
硅烷偶联剂a-1核磁信息如下:
[0162]1hnmr(dmso):δ:0.87(t,4h),1.41(m,4h),1.64(m,4h),2.71(m,2h),2.90~3.10(m, 4h),3.83(s,18h),10.36(s,1h),12.3(s,2h),13.75(s,2h)。
[0163]
实施例2
[0164]
硅烷偶联剂a-2的合成:
[0165]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,3,5-二氨基-1,2,4-三氮唑9.91g(0.1mol,阿拉丁化学试剂),开启搅拌,待3,5
‑ꢀ
二氨基-1,2,4-三氮唑充分溶解后,缓慢加入3-(三甲氧基硅烷基)丙基丁二酸酐52.47 g(0.2mol,x-12-967c,信越化学),待加料完成后,25℃下继续反应20hr后向反应体系中加入吡啶31.64g(0.4mol),搅拌均匀后缓慢加入乙酸酐40.84g(0.4mol),25℃反应20hr,反应完成后,减压蒸馏,除去体系中的溶剂、乙酸酐和生成的乙酸、吡啶,得到纯化的硅烷偶联剂a-2。
[0166][0167]
所得硅烷偶联剂a-2结构式如(a-2)所示。
[0168]
硅烷偶联剂a-2核磁信息如下:
[0169]1hnmr(dmso):δ:0.85(t,4h),1.45(m,4h),1.61(m,4h),2.72(m,2h),2.85~3.08(m, 4h),3.83(s,18h),10.2(s,1h)。
[0170]
实施例3
[0171]
硅烷偶联剂a-3的合成:
[0172]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,3,5-二氨基-1,2,4-三氮唑9.91g(0.1mol,阿拉丁化学试剂),开启搅拌,待3,5
‑ꢀ
二氨基-1,2,4-三氮唑充分溶解后,缓慢加入硅烷偶联剂sanah 65.28g(0.2mol),待加料完成后25℃下继续反应20hr,反应完成后进行减压蒸馏纯化得到硅烷偶联剂a-3。
[0173][0174]
所得硅烷偶联剂a-3结构式如(a-3)所示。
[0175]
硅烷偶联剂a-3核磁信息如下:
[0176]1hnmr(dmso):δ:0.85(t,4h),1.45(m,4h),1.6~1.71(m,4h),2.34(t,4h),2.66(m, 2h),2.81(t,2h),2.91(t,2h),3.8(m,2h),3.95(s,18h),5.94(d,2h),10.1(s,1h),12.3(s,2h), 13.5(s,2h)。
[0177]
实施例4
[0178]
硅烷偶联剂a-4的合成:
[0179]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,3,5-二氨基-1,2,4-三氮唑9.91g(0.1mol,阿拉丁化学试剂),开启搅拌,待3,5
‑ꢀ
二氨基-1,2,4-三氮唑充分溶解后,缓慢加入硅烷偶联剂sanah 65.28g(0.2mol),待加料完成后,25℃下继续反应20hr后向反应体系中加入吡啶31.64g(0.4mol),搅拌均匀后缓慢加入乙酸酐40.84g(0.4mol),25℃反应20hr,反应完成后,减压蒸馏,除去体系中的溶剂、乙酸酐和生成的乙酸、吡啶,得到纯化的硅烷偶联剂a-4。
[0180][0181]
所得硅烷偶联剂a-4结构式如(a-4)所示。
[0182]
硅烷偶联剂a-4核磁信息如下:
[0183]1hnmr(dmso):δ:0.85(t,4h),1.45(m,4h),1.6~1.73(m,4h),2.32(t,4h),2.66(m, 2h),3.66(m,4h),3.83(m,2h),3.99(s,18h),5.94(d,2h),10.1(s,1h)。
[0184]
实施例5
[0185]
二胺单体b的合成:
[0186]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,3,5-二氨基-1,2,4-三氮唑19.82g(0.2mol,阿拉丁化学试剂),搅拌溶解,待3,5
‑ꢀ
二氨基-1,2,4-三氮唑充分溶解后升温至80℃,缓慢加入21.81g(0.1mol,阿拉丁化学试剂)均苯四甲酸酐,80℃反应12h后25℃下继续反应20hr,然后向反应体系中加入吡啶31.64g(0.4mol),搅拌均匀后缓慢加入乙酸酐40.84g(0.4mol),25℃反应20hr,反应完成后,将所得到的反应液倒入水溶液中,析出聚合物沉淀,将所得到的沉淀物滤出后用去离子水洗涤三遍,在50℃下进行真空干燥72hr,得到二胺单体b。
[0187]
硅烷偶联剂a-5的合成:
[0188]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,二胺单体b 38.03g(0.1mol),开启搅拌,待二胺单体b充分溶解后,缓慢加入 3-(三甲氧基硅烷基)丙基丁二酸酐52.47g(0.2mol,x-12-967c,信越化学),待加料完成后25℃下继续反应20hr,反应完成后进行减压蒸馏纯化得到硅烷偶联剂a-5。
[0189][0190]
所得硅烷偶联剂a-5结构式如(a-5)所示。
[0191]
硅烷偶联剂a-5核磁信息如下:
[0192]1hnmr(dmso):δ:0.83(t,4h),1.43(m,4h),1.61(m,4h),2.75(m,2h),2.86~3.01(m, 4h),3.92(s,18h),8.65(s,2h),10.2(s,2h),12.3(s,2h),13.5(s,2h)。
[0193]
实施例6
[0194]
二胺单体b的合成:如前所述。
[0195]
硅烷偶联剂a-6的合成:
[0196]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,二胺单体b 38.03g(0.1mol),开启搅拌,待二胺单体b充分溶解后,缓慢加入 3-(三甲氧基硅烷基)丙基丁二酸酐52.47g(0.2mol,x-12-967c,信越化学),待加料完成后25℃下继续反应20hr,然后向反应体系中加入吡啶31.64g(0.4mol),搅拌均匀后缓慢加入乙酸酐40.84g(0.4mol),25℃反应20hr,反应完成后,减压蒸馏,除去体系中的溶剂、乙酸酐和生成的乙酸、吡啶,得到硅烷偶联剂a-6。
[0197][0198]
所得硅烷偶联剂a-6结构式如(a-6)所示。
[0199]
硅烷偶联剂a-6核磁信息如下:
[0200]1hnmr(dmso):δ:0.82(t,4h),1.43(m,4h),1.60(m,4h),2.80(m,2h),2.95~3.12(m, 4h),3.95(s,18h),8.65(s,2h),10.2(s,2h)。
[0201]
实施例7
[0202]
二胺单体b的合成:如前所述。
[0203]
硅烷偶联剂a-7的合成:
[0204]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,二胺单体b 38.03g(0.1mol),开启搅拌,待二胺单体b充分溶解后,缓慢加入硅烷偶联剂sanah 65.28g(0.2mol),待加料完成后25℃下继续反应20hr,反应完成后进行减压蒸馏纯化得到硅烷偶联剂a-7。
[0205][0206]
所得硅烷偶联剂a-7结构式如(a-7)所示。
[0207]
硅烷偶联剂a-7核磁信息如下:
[0208]1hnmr(dmso):δ:0.86(t,4h),1.45(m,4h),1.59~1.75(m,4h),2.34(t,4h),2.68(m, 2h),2.75~2.85(m,4h),3.65(m,2h),3.98(s,18h),5.88(d,2h),8.54(d,2h),10.3(s,2h), 12.1(s,2h),13.5(s,2h)。
[0209]
实施例8
[0210]
二胺单体b的合成:如前所述。
[0211]
硅烷偶联剂a-8的合成:
[0212]
向配有搅拌器和温度计的500ml三口烧瓶中依次加入溶剂n-甲基吡咯烷酮250 ml,二胺单体b 38.03g(0.1mol),开启搅拌,待二胺单体b充分溶解后,缓慢加入硅烷偶联剂sanah 65.28g(0.2mol),待加料完成后25℃下继续反应20hr,后向反应体系中加入吡啶31.64g(0.4mol),搅拌均匀后缓慢加入乙酸酐40.84g(0.4mol),25℃反应20hr,反应完成后,减压蒸馏,除去体系中的溶剂、乙酸酐和生成的乙酸、吡啶,得到硅烷偶联剂a-8。
[0213][0214]
所得硅烷偶联剂a-8结构式如(a-8)所示。
[0215]
硅烷偶联剂a-8核磁信息如下:
[0216]1hnmr(dmso):δ:0.86(t,4h),1.45(m,4h),1.6~1.74(m,4h),2.34(t,4h),2.68(m, 2h),3.63~3.68(m,6h),3.96(s,18h),5.88(d,2h),8.54(d,2h),10.2(s,2h)。
[0217]
树脂组合物的制备
[0218]
参考例1
[0219]
聚酰亚胺前体c-1的合成:
[0220]
将31.02g(0.1mol)的4,4
’‑
氧双邻苯二甲酸二酐(odpa)放入250ml的三口烧瓶中,加入26.03g(0.2mol)甲基丙烯酸羟乙酯(hema)和100mlγ-丁内酯(gbl),在10℃以下,边搅
拌边滴入(0.2mol)15.82g吡啶,得到反应混合物,自然升至25℃后搅拌12hr。
[0221]
接下来,在冰浴条件下,用40min边搅拌边将该反应混合物加入溶有41.25g (0.2mol)二环己基碳化二亚胺(dcc)的50ml的gbl溶液中,在氮气保护下,用 60min加入溶有19.03g(0.095mol)4,4
’‑
二氨基二苯基醚(oda)的70ml的gbl溶液,自然升至25℃,加入80ml gbl,继续搅拌12hr后加入乙醇6.0g并搅拌1hr,过滤除去反应混合物中产生的沉淀物,得到反应液。
[0222]
将所得到的反应液加入到1l的乙醇中,析出聚合物,再用300ml四氢呋喃将析出的聚合物溶解,将所得到的聚合物溶液滴入5l超纯水中,析出聚合物沉淀,将所得到的沉淀物滤出后,在50℃下进行真空干燥72hr,得到聚合物粉末,即聚酰亚胺前体 c-1。用凝胶渗透色谱法(标准聚苯乙烯换算)测定聚合物粉末的分子量,重均分子量 (mw)为1.8~2.2万。
[0223]
树脂组合物的制备:
[0224]
在配有搅拌的三口烧瓶中,将10.0g合成好的聚酰亚胺前体c-1溶解在20g n-甲基吡咯烷酮(nmp)中,待组分c-1完全溶解后,加入0.5g实施例1获得的硅烷偶联剂a-1,继续搅拌至完全溶解,再依次加入0.3g光引发剂1-(4-苯硫基-苯基)-辛-1, 2-二酮-2-肟-0-苯甲酸酯(oxe-1,basf)、1.5g交联剂三缩四乙二醇二甲基丙烯酸酯 (tegdma)、0.1g热阻聚剂对羟基苯甲醚(mehq),充分溶解后,利用1.0um滤膜压滤,得到负型感光性树脂组合物,25℃下测得粘度为3000cp。
[0225]
参考例2
[0226]
除了将硅烷偶联剂a-1由0.5g变为0.2g以外,其他同参考例1。
[0227]
参考例3
[0228]
除了将硅烷偶联剂a-1由0.5g变为0.1g以外,其他同参考例1。
[0229]
参考例4
[0230]
除了将硅烷偶联剂a-1由0.5g变为1.0g以外,其他同参考例1。
[0231]
参考例5
[0232]
除了将硅烷偶联剂a-1由0.5g变为2.0g以外,其他同参考例1。
[0233]
参考例6
[0234]
除了将硅烷偶联剂a-1变为硅烷偶联剂a-2以外,其他同参考例1。
[0235]
参考例7
[0236]
除了将硅烷偶联剂a-1变为硅烷偶联剂a-3以外,其他同参考例1。
[0237]
参考例8
[0238]
除了将硅烷偶联剂a-1变为硅烷偶联剂a-4以外,其他同参考例1。
[0239]
参考例9
[0240]
除了将硅烷偶联剂a-1变为硅烷偶联剂a-5以外,其他同参考例1。
[0241]
参考例10
[0242]
除了将硅烷偶联剂a-1变为硅烷偶联剂a-6以外,其他同参考例1。
[0243]
参考例11
[0244]
除了将硅烷偶联剂a-1变为硅烷偶联剂a-7以外,其他同参考例1。
[0245]
参考例12
[0246]
除了将硅烷偶联剂a-1变为硅烷偶联剂a-8以外,其他同参考例1。
[0247]
参考例13
[0248]
聚苯并噁唑前体的合成:
[0249]
在氮气流下,将36.57g(0.10mol)的2,2-双(3-氨基-4-羟基苯基)六氟丙烷(bahf)、 100g n-甲基吡咯烷酮(nmp)及52.8g(0.6mol)缩水甘油基甲醚加入到500ml三口烧瓶中,充分溶解后,将溶液的温度冷却至-15℃。向烧瓶里滴加29.4g(0.10mol) 二苯醚二甲酰氯溶解于50g n-甲基吡咯烷酮制成的溶液,滴加过程中控制反应物料在 0℃以下。滴加完成后,在-10~-15℃条件下,继续搅拌反应6小时,反应结束。将反应液倒入3l的10wt%甲醇水溶液中,析出聚合物得白色沉淀。过滤后用去离子水洗涤三次,放入真空烘箱中,50℃干燥72h,即得到聚苯并噁唑前体树脂c-2。用凝胶渗透色谱法(标准聚苯乙烯换算)测定聚合物粉末的分子量,结果重均分子量(mw)为 2.1~2.5万。
[0250]
树脂组合物的制备:
[0251]
在配有搅拌的三口烧瓶中,将10.0g合成好的聚苯并噁唑前体树脂c-2溶解在 20gn-甲基吡咯烷酮(nmp)中,搅拌至完全溶解,再依次加入0.5g实施例1获得的硅烷偶联剂a-1,2.0g感光剂醌二叠氮化合物nt-300(日本东洋合成工业(株)制造),充分溶解后,利用1.0um滤膜压滤,得到正型感光性树脂组合物,25℃下测得粘度为 3100cp。
[0252]
比较例1
[0253]
除了将硅烷偶联剂a-1变为0.41g 3-(三甲氧基硅烷基)丙基丁二酸酐(x-12-967c,信越化学)和0.09g苯并三唑以外,其它同参考例1。
[0254]
比较例2
[0255]
除了将硅烷偶联剂a-1变为0.41g硅烷偶联剂sanah和0.09g苯并三唑以外,其它同参考例1。
[0256]
比较例3
[0257]
除了将硅烷偶联剂a-1变为0.41g 3-(三甲氧基硅烷基)丙基丁二酸酐(x-12-967c,信越化学)和0.09g三氮唑以外,其它同参考例1。
[0258]
比较例4
[0259]
除了将硅烷偶联剂a-1变为0.41g 3-(三甲氧基硅烷基)丙基丁二酸酐(x-12-967c,信越化学)和0.09g8-氮杂鸟嘌呤以外,其它同参考例1。
[0260]
比较例5
[0261]
含三氮唑基团聚酰亚胺前体c-3的合成:
[0262]
将31.02g(0.1mol)的4,4
’‑
氧双邻苯二甲酸二酐(odpa)放入250ml的三口烧瓶中,加入26.03g(0.2mol)甲基丙烯酸羟乙酯(hema)和100mlγ-丁内酯(gbl),在10℃以下,边搅拌边滴入(0.2mol)15.82g吡啶,得到反应混合物,自然升至25℃后搅拌12hr。
[0263]
接下来,在冰浴条件下,用40min边搅拌边将该反应混合物加入溶有41.25g (0.2mol)二环己基碳化二亚胺(dcc)的50ml的gbl溶液中,在氮气保护下,用 60min加入溶有17.02g(0.085mol)4,4
’‑
二氨基二苯基醚(oda)和3,5-二氨基-1,2,4
‑ꢀ
三氮唑0.99g(0.01mol,阿拉丁化学试剂)的70ml的gbl溶液,自然升至25℃,加入80ml gbl,继续搅拌12hr后加入乙醇6.0g并搅拌1hr,过滤除去反应混合物中产生的沉淀物,得到反应液。
[0264]
将所得到的反应液加入到1l的乙醇中,析出聚合物,再用300ml四氢呋喃将析出的聚合物溶解,将所得到的聚合物溶液滴入5l超纯水中,析出聚合物沉淀,将所得到的沉淀物
滤出后,在50℃下进行真空干燥72hr,得到聚合物粉末,即含三氮唑基团聚酰亚胺前体c-3。用凝胶渗透色谱法(标准聚苯乙烯换算)测定聚合物粉末的分子量,结果重均分子量(mw)为1.8~2.2万。
[0265]
树脂组合物的制备:
[0266]
在配有搅拌的三口烧瓶中,将10.0g合成好的含三氮唑基团聚酰亚胺前体c-3溶解在20gn-甲基吡咯烷酮(nmp)中,搅拌至完全溶解,加入0.5g 3-(三甲氧基硅烷基)丙基丁二酸酐(x-12-967c,信越化学),继续搅拌至完全溶解后,再依次加入0.3g 光引发剂1-(4-苯硫基-苯基)-辛-1,2-二酮-2-肟-0-苯甲酸酯(oxe-1,basf)、1.5g 交联剂三缩四乙二醇二甲基丙烯酸酯(tegdma)、0.1g热阻聚剂对羟基苯甲醚 (mehq),充分溶解后,利用1.0um滤膜压滤,得到负型感光性树脂组合物,25℃下测得粘度为3100cp。
[0267]
比较例6
[0268]
除了不加硅烷偶联剂外,其它同参考例1。
[0269]
按照前面所述的粘合性剥离试验方法和铜变色试验对上述制备的树脂组合物进行评价,结果见表1及图4。
[0270]
从表1参考例及比较例可以看出,本发明硅烷偶联剂a-1~a-8对聚酰亚胺类树脂与铜基板的粘合性能均有较好的提升作用,即使在pct老化处理后依然表现出对铜基板的高粘附力。硅烷偶联剂a-1~a-6在提升树脂与铜基板的粘合性方面,以及抑制铜基板变色方面效果相当。a-7~a-8在增强树脂与铜基板的粘合性方面效果略差,可能是由于二者分子量较大,含有的硅氧烷基团密度偏低导致。
[0271]
在硅烷偶联剂的用量为耐热性树脂质量的1~10%的范围内,树脂固化后与基材均有较好的粘合性,因此硅烷偶联剂的用量优选为耐热性树脂质量的1~10%。
[0272]
从比较例1~3看,硅烷偶联剂x-12-967c和苯并三唑或三氮唑机械混合后,虽然也有提升树脂与铜基板的粘合性能的作用,但是其提升效果明显低于本发明;同时,其抑制铜或铜合金变色的效果也明显低于本发明。
[0273]
从比较例4看,将硅烷偶联剂x-12-967c和8-氮杂鸟嘌呤机械混合加入树脂组合物,有较好的抑制铜基板变色的效果,但是其增强树脂对铜基板粘结性的效果明显低于本发明。
[0274]
从比较例5看,相较于在树脂中另外添加三氮唑基化合物,直接在树脂结构中引入三氮唑基团,使得树脂组合物中有较多的三氮唑基团,因此有较好的抑制铜基板变色的效果,但是该比较例中树脂与铜基板粘结性的效果明显低于本发明。
[0275]
图4为比较例6(图4a)及参考例1(图4b)中,rdl与树脂固化膜结合界面的扫描电镜图片。从图4中可以看出,加入本发明中的硅烷偶联剂a-1后,树脂固化膜与rdl的粘结界面无剥离。
[0276]
本发明的硅烷偶联剂可显著增强聚酰亚胺及聚苯并噁唑等耐热性树脂在铜或铜合金基材上高温热固化后与基材的粘合性,同时可显著改善铜或铜合金基材的变色问题。
[0277]
[0278]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。