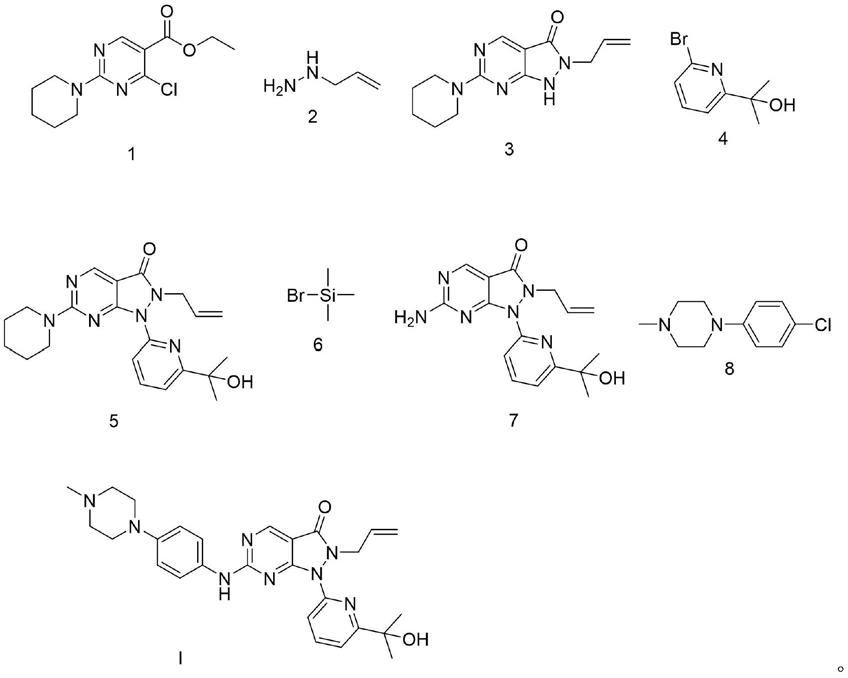
wee1抑制剂adavosertib的制备工艺
技术领域
1.本发明涉及生物医药技术领域,具体地,涉及一种wee1抑制剂adavosertib的制备 工艺。
背景技术:
2.阿斯利康公司经默克公司许可,正在开发一种wee
‑
1蛋白激酶抑制剂adavosertib(研 发代号azd
‑
1775;mk
‑
1775),其可用于口服治疗实体瘤。adavosertib是一种小分子抑制 剂,剂型为胶囊。2012年4月,欧洲ema授予adavosertib治疗卵巢癌的孤儿药物称号。 adavosertib目前在美国、加拿大、欧洲、荷兰、韩国最高处于临床2期试验阶段。其处 于2期临床的适应症为:乳腺癌、输卵管癌、头颈部肿瘤、卵巢癌、胰腺癌、腹膜肿瘤、 小细胞肺癌、子宫肿瘤;其在美国、澳大利亚、加拿大、法国、日本、韩国、西班牙、 英国有处于1期临床研究,1期临床的适应症为:晚期实体瘤、转移性膀胱癌、小细胞肺 癌、实体瘤。
3.adavosertib的化学结构在wo2011034743中披露,其结构式具体如式i所示化合物:
[0004][0005]
然而,目前adavosertib的化学合成制备工艺仍有待改进。
技术实现要素:
[0006]
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一 个目的在于提出一种新的wee1抑制剂adavosertib的制备工艺。相对于现有技术,本发 明所述的制备工艺,有效提高了反应的总收率和工业化操作性。
[0007]
在本发明的一个方面,本发明提供了一种式i所示化合物adavosertib的制备工艺。 根据本发明的实施例,该制备工艺包括:
[0008]
(1)使式1所示化合物与式2所示化合物接触,以便获得式3所示化合物;
[0009]
(2)使式3所示化合物与式4所示化合物接触,以便获得式5所示化合物;
[0010]
(3)使式5所示化合物与式6所示化合物接触,以便获得式7所示化合物;
[0011]
(4)使式7所示化合物与式8所示化合物接触,以便获得式i所示化合物,
[0012][0013]
发明人发现,利用本发明所述的制备工艺,采用已有的中间体,经过微波反应、卤 代反应、氨基脱保护后,再经卤代反应,能够快速、有效地制备获得式i所示化合物 adavosertib。
[0014]
在本文中所使用的术语“接触”应做广义理解,其可以是任何能够使得至少两种反应 物发生化学反应的方式,例如可以是将两种反应物在适当的条件下进行混合。根据需要, 可以在搅拌下,将需要进行接触的反应物进行混合,由此,搅拌的类型并不受特别限制, 例如可以为机械搅拌,即在机械力的作用下进行搅拌。
[0015]
在本文中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要 性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明 示或者隐含地包括一个或者更多个该特征。在本发明的描述中,“多个”的含义是两个或 两个以上,除非另有明确具体的限定。
[0016]
根据本发明的实施例,上述制备式3所示化合物、式5所示化合物、式7所示化合 物、式i所示化合物的方法还可以具有下列附加技术特征至少之一:
[0017]
根据本发明的实施例,本发明所述的化学反应可以按照本领域已知的任何方法进行。 制备式3所示化合物、式5所示化合物、式7所示化合物、式i所示化合物的原料的来 源并不受特别限制,其可以是采用任何已知的方法制备的,或者市售获得的。
[0018]
根据本发明的实施例,在步骤(1)中,式1所示化合物与式2所示化合物的接触方 式并不受特别限制。由此,可以提升式1所示化合物与式1所示化合物接触反应的效率, 加快微波反应的速度,进一步提高利用该方法制备式3所示化合物的效率。
[0019]
根据本发明的实施例,在步骤(1)中,包括如下步骤:将式1所示化合物和式2所 示
化合物混合,在没有溶剂的情况下,使用微波管进行反应,反应结束后向反应混合物 中加水,过滤收集固体产品,将固体用含c1
‑
c5的直链或支链醇打浆纯化得到式3所示 化合物。由此,可以提升式1所示化合物与式2所示化合物接触反应的效率,加快微波 反应的速度,进一步提高利用该方法制备式3所示化合物的效率。
[0020]
根据本发明的实施例,在步骤(1)中,式1所示化合物与式2所示化合物的摩尔比 为1:(1.05~1.2),优选式1所示化合物与式2所示化合物的摩尔比为1:1.1。由此,反 应物利用率较高,不会造成原料、实际的浪费,目标化合物收率较高,可以进一步提高 利用该方法制备式3所示化合物的效率。
[0021]
根据本发明的实施例,在步骤(1)中,式1所示化合物与肼进行微波反应,在室温 功率为400w下,反应5~6分钟,由此,可以提升式1所示化合物与肼进行微波反应的 效率,进一步提高利用该方法制备式3所示化合物的效率。
[0022]
根据本发明的实施例,在步骤(1)中,所述含c1
‑
c5的直链或支链醇选自甲醇、乙 醇、异丙醇、丁醇、戊醇。
[0023]
根据本发明的一个具体实施例,在步骤(1)中,包括如下步骤:20℃下,将27.0g 式1所示化合物和7.92g式2所示化合物混合,在没有溶剂的情况下,使用微波管进行反 应,在室温功率为400w下反应5分钟,反应结束后向反应混合物中加入30毫升水,过 滤收集固体产品,将固体用120ml甲醇打浆纯化,过滤,固体干燥后得到式3所示化 合物,得量24.9g,收率96.0%。
[0024]
根据本发明的实施例,在步骤(2)中,式3所示化合物、k2co3、与式4所示化合 物的接触方式并不受特别限制。由此,可以提升式3所示化合物、k2co3、与式4所示化 合物接触反应的效率,加快反应速度,进一步提高利用该方法制备式5所示化合物的效 率。
[0025]
根据本发明的实施例,在步骤(2)中,包括如下步骤:室温下,将式3所示化合物 和k2co3加入搅拌中的dmf中混合,再加入式4所示化合物,升温搅拌反应,反应毕, 进行后处理及打浆纯化,过滤固体,干燥后得到式5所示化合物。由此,可以提升式3 所示化合物、k2co3、与式4所示化合物接触反应的效率,加快反应速度,进一步提高利 用该方法制备式5所示化合物的效率。
[0026]
根据本发明的实施例,在步骤(2)中,式3所示化合物、k2co3、式4所示化合物 的摩尔比为1:(1.0~1.2):(1.0~1.2),优选式3所示化合物、k2co3、式4所示化合物 的摩尔比为1:1.1:1.05。由此,可以进一步提高利用该方法制备式5所示化合物的效率。
[0027]
根据本发明的实施例,在步骤(2)中,式3所示化合物、k2co3、式4所示化合物 接触搅拌的反应时间为1.5~2.5h,优选式3所示化合物、k2co3、式4所示化合物接触搅 拌的反应时间为2小时。由此,可以提升式3所示化合物、k2co3、式4所示化合物接触 反应的效率,进一步提高利用该方法制备式5所示化合物的效率。
[0028]
根据本发明的实施例,在步骤(2)中,式3所示化合物、k2co3、式4所示化合物 接触搅拌升温的反应温度为55℃~65℃,优选式3所示化合物、k2co3、式4所示化合物 升温至60℃搅拌反应。由此,可以提升式3所示化合物、k2co3、式4所示化合物接触 反应的效率,进一步提高利用该方法制备式5所示化合物的效率。
[0029]
根据本发明的实施例,在步骤(2)中,所述打浆纯化采用体积比为1:1的石油醚/ 乙酸乙酯的混合溶剂。
[0030]
根据本发明的一个具体实施例,在步骤(2)中,包括如下步骤:室温下,将5.19g 式3所示化合物和3.04g k2co3加入搅拌中的40mldmf中混合,再加入4.54g式4所 示化合物,升温至60℃搅拌反应2h,反应毕,将反应液倒入40ml水中,将析出的固体 过滤收集后,用体积比为1:1的石油醚/乙酸乙酯组成的50ml混合溶剂搅拌打浆纯化 30min,过滤固体,干燥后得到式5所示化合物,得量7.22g,收率91.5%。
[0031]
根据本发明的实施例,在步骤(3)中,式5所示化合物与式6所示化合物的接触方 式并不受特别限制。由此,可以提升式5所示化合物与式6所示化合物接触反应的效率, 加快反应速度,进一步提高利用该方法制备式7所示化合物的效率。
[0032]
根据本发明的实施例,在步骤(3)中,包括如下步骤:室温下,将式5所示化合物 和式6所示化合物加入二氯甲烷中,反应液搅拌升温回流反应,反应毕,将反应液降至 室温,加入冰水搅拌,再加入饱和nahco3溶液洗涤后,有机层减压浓缩至干,将粗产 物用混合溶剂搅拌打浆纯化,过滤固体,干燥后得到式7所示化合物。由此,可以提升 式5所示化合物与式6所示化合物接触反应的效率,加快反应速度,进一步提高利用该 方法制备式7所示化合物的效率。
[0033]
根据本发明的实施例,在步骤(3)中,式5所示化合物与式6所示化合物的摩尔比 为1:(8~12),优选式5所示化合物与式6所示化合物的摩尔比为1:10。由此,反应物 利用率较高,不会造成原料、实际的浪费,目标化合物收率较高。
[0034]
根据本发明的实施例,在步骤(3)中,式5所示化合物与式6所示化合物接触搅拌 的反应时间为3.5小时~5小时,优选式5所示化合物与式6所示化合物搅拌接触的反应 时间为4小时。由此,可以提升式5所示化合物与式6所示化合物接触反应的效率,进 一步提高利用该方法制备式7所示化合物的效率。
[0035]
根据本发明的实施例,在步骤(3)中,所述打浆纯化采用体积比为1:1的石油醚/ 乙酸乙酯的混合溶剂。
[0036]
根据本发明的一个具体实施例,在步骤(3)中,包括如下步骤:室温下,将3.94g 式5所示化合物和15.3g式6所示化合物加入60ml二氯甲烷中,反应液搅拌升温回流 反应4个小时,反应毕,将反应液降至室温,加入30ml冰水搅拌,再加入30ml饱和 nahco3溶液洗涤后,有机层减压浓缩至干,将粗产物用体积比为1:1的石油醚/乙酸乙 酯组成的混合溶剂30ml搅拌打浆纯化30min,过滤固体,干燥后得到式7所示化合物, 得量2.58g,收率79.1%。
[0037]
根据本发明的实施例,在步骤(4)中,式7所示化合物、k2co3、式8所示化合物 的接触方式并不受特别限制。由此,可以提升式7所示化合物、k2co3、式8所示化合物 接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物的效率。
[0038]
根据本发明的实施例,在步骤(4)中,包括如下步骤:室温下,将式7所示化合物 和k2co3加入搅拌中的dmf中,再加入式8所示化合物,将混合物升温搅拌反应,反 应毕,将反应液降至室温后倒入水中,用乙酸乙酯萃取混合物,合并有机相后用饱和食 盐水洗涤后,硫酸钠干燥,过滤,滤液浓缩后采用硅胶柱层析纯化得式i所示化合物 adavosertib。由此,可以进一步提高利用该方法制备式i所示化合物adavosertib的效率。
[0039]
根据本发明的实施例,在步骤(4)中,式7所示化合物、k2co3、式8所示化合物 的摩尔比为1:(1.0~1.3):(1.0~1.2),优选式7所示化合物、k2co3、式8所示化合物 的摩尔比为1:1.15:1.05。由此,反应物利用率较高,不会造成原料、实际的浪费,目标 化合物收率较
高。
[0040]
根据本发明的实施例,在步骤(4)中,式7所示化合物、k2co3、式8所示化合物 接触搅拌的反应时间为45分钟~1.5h,优选式7所示化合物、k2co3、式8所示化合物接 触搅拌的反应时间为1小时。由此,可以提升式7所示化合物、k2co3、式8所示化合物 接触反应的效率,进一步提高利用该方法制备式i所示化合物的效率。
[0041]
根据本发明的实施例,在步骤(4)中,式7所示化合物、k2co3、式8所示化合物 接触搅拌升温的反应温度为55℃~65℃,优选式7所示化合物、k2co3、式8所示化合物 升温至60℃搅拌反应。由此,可以提升式7所示化合物、k2co3、式8所示化合物接触 反应的效率,进一步提高利用该方法制备式i所示化合物的效率。
[0042]
根据本发明的实施例,在步骤(4)中,所述硅胶柱层析采用体积比为(10~30):1 的二氯甲烷/甲醇的混合溶剂,优选柱层析采用体积比为20:1的二氯甲烷/甲醇的混合溶 剂。
[0043]
根据本发明的一个具体实施例,在步骤(4)中,包括如下步骤:室温下,将32.6g 式7所示化合物和15.9g k2co3加入搅拌中的200mldmf中,再加入22.1g式8所示化 合物,将混合物升温至60℃搅拌反应1h,反应毕,将反应液降至室温后倒入40ml水 中,用3
×
40ml乙酸乙酯萃取混合物,合并有机相后用30ml饱和食盐水洗涤后,硫酸 钠干燥,过滤,滤液浓缩后采用体积比为20:1的二氯甲烷/甲醇的混合溶剂进行硅胶柱 层析纯化得式i所示化合物adavosertib,固体得量44.0g,收率87.9%,hplc纯度99.5%。
[0044]
根据本发明的实施例,式i所示化合物adavosertib的制备工艺的合成路线可以如下 所示:
[0045][0046]
相对于现有技术,本发明所述的wee1抑制剂adavosertib的制备工艺,其至少具有 以下有益效果:本路线采用已有的中间体,经过微波反应、卤代反应、氨基脱保护后, 再经卤代反应合成目标化合物adavosertib。更具体地,本发明的步骤(1)选择氨基保护 的式1所示化合物为原料,利用微波反应关环可以大幅度减少反应时间,提高反应产率; 步骤(2)中的卤代反应选择价格低廉的k2co3作为碱性试剂参与反应,避免了使用价格 昂贵的nah,降低了反应成本;步骤(3)利用式6所示化合物(溴甲基苯丙烷)脱去式 5所示化合物中的氨基保护基;步骤(4)采用k2co3作为碱性试剂参与反应,可以特异 性的进行式7所示化合物中氨基的卤代反应,减少式7所示化合物中
‑
oh卤代副产物的 生成。本发明所述的制备工艺,其反应步骤少、各个步骤的反应难度较小,反应后处理 操作简单。此外,本发明利用产物溶解性较差的特点,反应的纯化大多采用打浆纯化的 方式,纯化过程简单、快速,总体反应产率更高。本发明所述的wee1抑制剂adavosertib 的制备工艺,有效提高了反应的总收率和工业化操作性。
具体实施方式
[0047]
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明, 而不能理解为对本发明的限制。实施例中未注明具体技术或条件的,按照本领域内的文 献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者, 均为可以通过市购获得的常规产品。
[0048]
实施例1式3所示化合物的合成
[0049]
20℃下,将式1所示化合物(27.0g,0.1mol)和式2所示化合物(7.92g,0.11mol)混合, 在没有溶剂的情况下,使用微波管进行反应,在室温功率为400w下反应5分钟,反应 结束后向反应混合物中加入水(30毫升),过滤收集固体产品,将固体用甲醇(120ml) 打浆纯化,过滤,固体干燥后得到式3所示化合物,得量24.9g,收率96.0%。
[0050]
lc
‑
ms(apci):m/z=260.3(m 1)
。
[0051]
实施例2式3所示化合物的合成
[0052]
20℃下,将式1所示化合物(27.0g,0.1mol)和式2所示化合物(7.57g,0.105mol)混合, 在没有溶剂的情况下,使用微波管进行反应,在室温功率为400w下反应5.5分钟,反应 结束后向反应混合物中加入水(5毫升),过滤收集固体产品,将固体用甲醇(20ml)打 浆纯化,过滤,固体干燥后得到式3所示化合物,得量24.3g,收率93.7%。
[0053]
实施例3式3所示化合物的合成
[0054]
20℃下,将式1所示化合物(27.0g,0.1mol)和式2所示化合物(8.65g,0.12mol)混合, 在没有溶剂的情况下,使用微波管进行反应,在室温功率为400w下反应6分钟,反应 结束后向反应混合物中加入水(5毫升),过滤收集固体产品,将固体用甲醇(20ml)打 浆纯化,过滤,固体干燥后得到式3所示化合物,得量24.4g,收率94.1%。
[0055]
实施例4式5所示化合物的合成
[0056]
室温下,将式3所示化合物(5.19g,20mmol)和k2co3(3.04g,22mmol)加入搅拌 中的dmf(40ml)中混合,再加入式4所示化合物(4.54g,21mmol),升温至60℃搅 拌反应2h,反应毕,将反应液倒入水中(40ml),将析出的固体过滤收集后,用体积 比为1:1的石油醚/乙酸乙酯组成的混合溶剂(50ml)搅拌打浆纯化30min,过滤固体, 干燥后得到式5所示化合物,得量7.22g,收率91.5%,产物hplc纯度为97.22%。
[0057]
lc
‑
ms(apci):m/z=395.3(m 1)
。
[0058]
实施例5式5所示化合物的合成
[0059]
室温下,将式3所示化合物(5.19g,20mmol)和k2co3(2.76g,20mmol)加入搅拌 中的dmf(40ml)中混合,再加入式4所示化合物(4.32g,20mmol),升温至55℃搅 拌反应2.5h,反应毕,将反应液倒入水中(40ml),将析出的固体过滤收集后,用体积 比为1:1的石油醚/乙酸乙酯组成的混合溶剂(50ml)搅拌打浆纯化30min,过滤固体, 干燥后得到式5所示化合物,得量7.18g,收率91.0%,hplc纯度为96.93%。
[0060]
实施例6式5所示化合物的合成
[0061]
室温下,将式3所示化合物(3.50g,20mmol)和k2co3(3.32g,24mmol)加入搅拌 中的dmf(40ml)中混合,再加入式4所示化合物(5.19g,24mmol),升温至65℃搅 拌反应1.5h,反应毕,将反应液倒入水中(40ml),将析出的固体过滤收集后,用体积 比为1:1的石油醚/乙酸乙酯组成的混合溶剂(50ml)搅拌打浆纯化30min,过滤固体, 干燥后得到式5所示化合物,得量7.08g,收率89.7%。
[0062]
实施例7式5所示化合物的合成
[0063]
实施例7为对比实施例,在本实施例7中,发明人调节式3所示化合物、k2co3、与 式4所示化合物的反应摩尔比为1:1.5:1.3,其技术效果上,得到的产物收率低于式3所 示化合物、k2co3、式4所示化合物的摩尔比为1:(1.0~1.2):(1.0~1.2)时的产物收率。
[0064]
室温下,将式3所示化合物(5.19g,20mmol)和k2co3(4.15g,30mmol)加入搅拌 中的
dmf(50ml)中混合,再加入式4所示化合物(5.62g,26mmol),升温至60℃搅 拌反应2h,反应毕,将反应液倒入水中(50ml),将析出的固体过滤收集后,用体积 比为1:1的石油醚/乙酸乙酯组成的混合溶剂(60ml)搅拌打浆纯化30min,过滤固体, 干燥后得到式5所示化合物,得量6.38g,收率80.9%,hplc纯度为91.60%。
[0065]
实施例8式5所示化合物的合成
[0066]
实施例8为对比实施例,在本实施例8中,发明人调节式3所示化合物、k2co3、与 式4所示化合物的反应摩尔比为1:0.95:0.98,其技术效果上,得到的产物收率低于式3 所示化合物、k2co3、式4所示化合物的摩尔比为1:(1.0~1.2):(1.0~1.2)时的产物收 率,中间体产物的纯度明显降低。
[0067]
室温下,将式3所示化合物(5.19g,20mmol)和k2co3(2.63g,19mmol)加入搅拌 中的dmf(40ml)中混合,再加入式4所示化合物(4.24g,19.6mmol),升温至60℃ 搅拌反应2h,反应毕,将反应液倒入水中(40ml),将析出的固体过滤收集后,用体 积比为1:1的石油醚/乙酸乙酯组成的混合溶剂(50ml)搅拌打浆纯化30min,过滤固体, 干燥后得到式5所示化合物,得量5.62g,收率71.3%,hplc纯度为85.54%。
[0068]
实施例9式6所示化合物的合成
[0069]
室温下,将式5所示化合物(3.94g,10mmol)和式6所示化合物(15.3g,100mmol)加入 二氯甲烷(60ml)中,反应液搅拌升温回流反应4个小时。反应毕,将反应液降至室温, 加入冰水(30ml)搅拌,再加入饱和nahco3溶液(30ml)洗涤后,有机层减压浓缩 至干,将粗产物用体积比为1:1的石油醚/乙酸乙酯组成的混合溶剂(30ml)搅拌打浆纯 化30min,过滤固体,干燥后得到式7所示化合物,得量2.58g,收率79.1%。
[0070]
lc
‑
ms(apci):m/z=327.2(m 1)
。
[0071]
实施例10式6所示化合物的合成
[0072]
室温下,将式5所示化合物(3.94g,10mmol)和式6所示化合物(12.2g,80mmol)加入二 氯甲烷(60ml)中,反应液搅拌升温回流反应3.5个小时。反应毕,将反应液降至室温, 加入冰水(30ml)搅拌,再加入饱和nahco3溶液(30ml)洗涤后,有机层减压浓缩 至干,将粗产物用体积比为1:1的石油醚/乙酸乙酯组成的混合溶剂(30ml)搅拌打浆纯 化30min,过滤固体,干燥后得到式7所示化合物,得量2.51g,收率76.9%。
[0073]
实施例11式6所示化合物的合成
[0074]
室温下,将式5所示化合物(3.94g,10mmol)和式6所示化合物(18.4g,120mmol)加入 二氯甲烷(65ml)中,反应液搅拌升温回流反应5个小时。反应毕,将反应液降至室温, 加入冰水(30ml)搅拌,再加入饱和nahco3溶液(30ml)洗涤后,有机层减压浓缩 至干,将粗产物用体积比为1:1的石油醚/乙酸乙酯组成的混合溶剂(30ml)搅拌打浆纯 化30min,过滤固体,干燥后得到式7所示化合物,得量2.55g,收率78.1%。
[0075]
实施例12式6所示化合物的合成
[0076]
实施例12为对比实施例,在本实施例中,发明人调节式5所示化合物与式6所示化 合物的反应摩尔比为1:6,反应时间为3.5小时。本对比例在技术效果上,得到的产物收 率低于式5所示化合物与式6所示化合物的摩尔比为1:(8~12)时的产物收率。
[0077]
室温下,将式5所示化合物(3.94g,10mmol)和式6所示化合物(9.2g,60mmol)加入二 氯甲烷(60ml)中,反应液搅拌升温回流反应3.5个小时。反应毕,将反应液降至室温, 加
入冰水(30ml)搅拌,再加入饱和nahco3溶液(30ml)洗涤后,有机层减压浓缩 至干,将粗产物用体积比为1:1的石油醚/乙酸乙酯组成的混合溶剂(30ml)搅拌打浆纯 化30min,过滤固体,干燥后得到式7所示化合物,得量2.07g,收率63.5%。
[0078]
实施例13式6所示化合物的合成
[0079]
实施例13为对比实施例,在本实施例中,发明人调节式5所示化合物与式6所示化 合物的反应摩尔比为1:15,反应时间为5小时。本对比例在技术效果上,得到的产物收 率低于式5所示化合物与式6所示化合物的摩尔比为1:(8~12)时的产物收率。
[0080]
室温下,将式5所示化合物(3.94g,10mmol)和式6所示化合物(23.0g,150mmol)加入 二氯甲烷(80ml)中,反应液搅拌升温回流反应5个小时。反应毕,将反应液降至室温, 加入冰水(40ml)搅拌,再加入饱和nahco3溶液(40ml)洗涤后,有机层减压浓缩 至干,将粗产物用体积比为1:1的石油醚/乙酸乙酯组成的混合溶剂(40ml)搅拌打浆纯 化30min,过滤固体,干燥后得到式7所示化合物,得量2.24g,收率68.7%。
[0081]
实施例14式i所示化合物adavosertib的合成
[0082]
室温下,将式7所示化合物(32.6g,0.1mol)和k2co3(15.9g,0.115mol)加入搅拌中的 dmf(200ml)中,再加入式8所示化合物(22.1g,0.105mol),将混合物升温至60℃搅 拌反应1h,反应毕,将反应液降至室温后倒入水中(40ml),用乙酸乙酯(3
×
40ml)萃 取混合物,合并有机相后用饱和食盐水(30ml)洗涤后,硫酸钠干燥,过滤,滤液浓缩后 采用体积比为20:1的二氯甲烷/甲醇的混合溶剂进行硅胶柱层析纯化得式i所示化合物 adavosertib,固体得量44.0g,收率87.9%,hplc纯度99.5%。
[0083]
lc
‑
ms(apci):m/z=501.4(m 1)
。
[0084]
实施例15式i所示化合物adavosertib的合成
[0085]
室温下,将式7所示化合物(32.6g,0.1mol)和k2co3(13.8g,0.1mol)加入搅拌中的 dmf(200ml)中,再加入式8所示化合物(21.1g,0.1mol),将混合物升温至65℃搅拌 反应45分钟,反应毕,将反应液降至室温后倒入水中(40ml),用乙酸乙酯(3
×
40ml) 萃取混合物,合并有机相后用饱和食盐水(30ml)洗涤后,硫酸钠干燥,过滤,滤液浓缩 后采用体积比为10:1的二氯甲烷/甲醇的混合溶剂进行硅胶柱层析纯化得式i所示化合 物adavosertib,固体得量42.4g,收率84.7%,hplc纯度99.2%。
[0086]
实施例16式i所示化合物adavosertib的合成
[0087]
室温下,将式7所示化合物(32.6g,0.1mol)和k2co3(18.0g,0.13mol)加入搅拌中的 dmf(200ml)中,再加入式8所示化合物(25.3g,0.12mol),将混合物升温至55℃搅拌 反应1.5h,反应毕,将反应液降至室温后倒入水中(40ml),用乙酸乙酯(3
×
40ml)萃 取混合物,合并有机相后用饱和食盐水(30ml)洗涤后,硫酸钠干燥,过滤,滤液浓缩后 采用体积比为30:1的二氯甲烷/甲醇的混合溶剂进行硅胶柱层析纯化得式i所示化合物 adavosertib,固体得量43.2g,收率86.3%,hplc纯度99.3%。
[0088]
实施例17式i所示化合物adavosertib的合成
[0089]
实施例17为对比实施例,在本实施例中,发明人调节式7所示化合物、k2co3、式 8所示化合物的摩尔比为1:1.5:1.5,其在技术效果上,得到的产物收率低于式7所示化 合物、k2co3、式8所示化合物的摩尔比为1:(1.0~1.3):(1.0~1.2)时的产物收率,且 得到的adavosertib化合物的终产物的纯度降低、杂质增多。
[0090]
室温下,将式7所示化合物(32.6g,0.1mol)和k2co3(20.7g,0.15mol)加入搅拌中的 dmf(250ml)中,再加入式8所示化合物(31.6g,0.15mol),将混合物升温至60℃搅拌 反应1.5h,反应毕,将反应液降至室温后倒入水中(40ml),用乙酸乙酯(3
×
40ml)萃 取混合物,合并有机相后用饱和食盐水(30ml)洗涤后,硫酸钠干燥,过滤,滤液浓缩后 采用体积比为30:1的二氯甲烷/甲醇的混合溶剂进行硅胶柱层析纯化得式i所示化合物 adavosertib,固体得量40.4g,收率80.8%,hplc纯度97.2%。
[0091]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、 或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点 包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不 必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以 在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本 领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特 征进行结合和组合。
[0092]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性 的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述 实施例进行变化、修改、替换和变型。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。