1.本发明属于化工领域,具体涉及一种具备工业适应性的以棒状二氧化铈催化2-氨基-4-甲硫基丁腈制备蛋氨酸的方法。
背景技术:
2.蛋氨酸(met,学名2-氨基-4-甲硫基丁酸)的化学法合成目前主要通过甲硫基丙醛与氢氰酸合成2-羟基-4-甲硫基丁腈(简称氰醇),氰醇再与二氧化碳、氨合成海因中间体,海因中间体经无机碱水解后得到蛋氨酸碱盐,酸化蛋氨酸碱盐后获得蛋氨酸;或甲硫基丙醛直接与氰化钠、二氧化碳、氨等组分合成海因中间体,海因中间体经碱水解、酸化后制得蛋氨酸。以上工艺虽已十分成熟,收率也较高,但仍存在无论采用何种碱盐水解海因,均会生成大量不必要的副产盐如硫酸钠、氯化钠等,且产物与副产盐分离困难,即使采用碳酸钾可实现水解、结晶、再循环水解的方式,大量碱盐存在体系中既会进一步导致体系内杂质形成和累积,同时也导致蛋氨酸单次取晶率较低,降低了生产效率。
3.中国专利201580051133.4公开了在含铈的氧化物催化剂存在下使2-氨基-4-甲硫基丁腈与水接触制备蛋氨酸的方法。具体的,该方法使用的含铈的氧化物催化剂选自三氧化二铈(ce2o3)、二氧化铈(ceo2)、它们的混合物或者具有它们混合相的氧化铈化合物以及含有铈的氧化物固溶体,所述固溶体可选自二氧化铈-氧化锆(ceo
2-zro2)、二氧化铈-氧化钇(ceo
2-y2o3)、二氧化铈-氧化镧(ceo
2-la2o3)等。该方法使2-氨基-4-甲硫基丁腈在含铈的氧化物催化剂存在下与水接触,反应结束得到蛋氨酸铵盐为主的反应料液,经浓缩得到蛋氨酸水溶液或蛋氨酸固体,分离出的催化剂可以循环利用或者再生,不产生大量副产盐,相比传统工艺具有一定优势。但其仍存在不足之处,首先,用该专利方法制备蛋氨酸的产率不稳定,从实施例中可以看到产率在较大幅度范围内变动;其次,所使用的氧化铈催化剂优选20~30nm(比表面积100m2/g以上)虽然有助于使单位质量催化剂具备更高的催化活性,但在实际应用中,难以与反应体系高效分离,且循环催化至3-5次催化效率即显著降低,外观由类白或淡黄色转为棕褐色,难以实现长期的循环催化;再次,其采用常压浓缩、减压浓缩等方式对主要成分为蛋氨酸铵盐的反应料液进行脱氨处理以获得蛋氨酸,脱氨处理方式耗时长且难以脱氨彻底,会导致体系中原有的杂质和蛋氨酸发生一些不必要的副反应使体系颜色加深和蛋氨酸难以完全取出,杂质与仍有相当一部分蛋氨酸会随着结晶母液通过循环进入体系,若返回催化阶段,会进一步加剧催化剂的性能降低和分离难度。
4.综上,现有技术存在以下主要问题:传统生产会生成大量不必要的副产盐,且蛋氨酸产物与副产盐分离困难;而使用氧化铈催化2-氨基-4-甲硫基丁腈制备蛋氨酸的工艺又存在反应时间过长、产率不稳定、难以脱氨彻底、不能实现催化剂长期循环催化的问题;另外,氧化铈具有球状、八面体、棒状、方块等多种晶型,不同晶型其暴露的活性面不同,晶型对氧化铈催化剂的性能具有重要影响,而现有工艺为了更显著利用氧化铈催化活性直接选择较小粒径和较大表面积的的氧化铈颗粒催化剂,这加剧了其应用缺陷,以上尺寸的小颗粒型或小球状催化剂更容易被聚合物、副反应物、色素杂质等吸附包裹污染造成活性降低,
且在污染后形成一定的聚合、絮凝效果,难以高效分离,催化剂滤饼往往含水高达30~50w%。
技术实现要素:
5.有鉴于此,本发明提供了一种蛋氨酸的制备方法,该方法制备的蛋氨酸取出率更高,二氧化铈催化剂可循环使用周期更长,本发明工艺具备工业适应性。
6.所述方法包括使ceo2固体催化剂与2-氨基-4-甲硫基丁腈含水液流接触的工序,所述ceo2固体催化剂为棒状晶型,晶体直径为10-80nm,长为100-500nm。
7.优选的,所述ceo2固体催化剂棒状晶体直径为20-60nm,更优选为30-50nm。
8.优选的,所述ceo2固体催化剂棒状晶体长为100-400nm,更优选为100-300nm。
9.优选的,所述ceo2固体催化剂棒状晶体的表面积为20-100cm2/g,更优选为30-80cm2/g。
10.优选的,所述2-氨基-4-甲硫基丁腈含水液流由2-氨基-4-甲硫基丁腈与补水液流混合制备得到,其中,2-氨基-4-甲硫基丁腈的含量为3-70w%。
11.优选的,2-氨基-4-甲硫基丁腈含水液流中2-氨基-4-甲硫基丁腈的含量为5-50w%,更优选为10-30w%
12.优选的,所述2-氨基-4-甲硫基丁腈由2-羟基-4-甲硫基丁腈与氨反应制得。
13.优选的,所述制备方法具体包括以下步骤:
14.(1)使所述ceo2固体催化剂与2-氨基-4-甲硫基丁腈含水液流接触,酰胺化反应制得蛋氨酸酰胺反应体系;
15.(2)对所述蛋氨酸酰胺反应体系进行非完全分离处理分别得到蛋氨酸酰胺水解液与催化剂体系;
16.(3)使所述蛋氨酸酰胺水解液与固体酸接触,经水解反应制得蛋氨酸水解液。
17.在本发明上下文中,所述蛋氨酸酰胺反应体系指所述ceo2固体催化剂与2-氨基-4-甲硫基丁腈接触进行酰胺化反应获得的包含水、ceo2固体催化剂、蛋氨酸酰胺的固液混合体系。
18.进一步的,所述蛋氨酸酰胺反应体系还包括蛋氨酸铵盐和/或蛋氨酸。
19.所述蛋氨酸酰胺水解液指从蛋氨酸酰胺反应体系中分离ceo2固体催化剂后获得的包含水、蛋氨酸酰胺的酰胺化反应获得的反应物溶液。
20.进一步的,所述蛋氨酸酰胺水解液还包括蛋氨酸铵盐和/或蛋氨酸。
21.所述催化剂体系指蛋氨酸酰胺反应体系经分离出蛋氨酸酰胺水解液后剩余的包含与所述蛋氨酸酰胺水解液成分一致的溶液和所述ceo2固体催化剂的固液混合体系。
22.优选的,所述蛋氨酸酰胺反应体系中,2-氨基-4-甲硫基丁腈的质量百分比≤0.05%。
23.优选的,步骤(2)中,所述非完全分离处理方式为静置沉降、倾析、滗析、离心、压滤、膜过滤中的一种或多种。
24.优选的,所述非完全分离处理方式为先静置倾析,再膜过滤。
25.优选的,所述过滤膜为超滤膜、微滤膜、陶瓷膜中的一种或多种。
26.优选的,将步骤(2)所得到的催化剂体系进行回收和/或在步骤(1)中进行利用,所
述催化剂体系包含与所述蛋氨酸酰胺水解液成分一致的溶液和所述ceo2固体催化剂,所述溶液满足在所述催化剂体系静置时至少覆盖所述ceo2固体催化剂的固体层;或所述溶液能够保证所述ceo2固体催化剂在一定外力作用下实现充分流动。
27.优选的,所述固体酸为氧化物固体酸、硫化物固体酸、金属盐固体酸、沸石分子筛固体酸、杂多酸固体酸、酸性阳离子交换树脂、固载化液体酸、固体超强酸中的一种或多种,所述酸性离子交换树脂为官能团为磺酸基、羧基、磷酸基、酚基的离子交换树脂中的一种或多种,所述固体超强酸为so42-/fe2o3、so42-/tio2、so42-/zro2、wo3/zro2、moo3/zro2中的一种或多种。
28.优选的,步骤(3)中,水解反应温度为60-300℃,更优选为80-200℃,更优选为90-160℃,
29.优选的,还包括对步骤(1)获得的蛋氨酸酰胺反应体系和/或步骤(2)获得的蛋氨酸酰胺水解液和/或对步骤(3)制得的蛋氨酸水解液进行脱氨处理的步骤。
30.优选的,所述脱氨处理方式为汽提、闪蒸、蒸馏中的一种或多种。
31.优选的,所述方法还包括步骤(4):向步骤(3)制得的蛋氨酸水解液中通入二氧化碳进行酸化反应制得结晶体系,分离所述结晶体系得到蛋氨酸固体和含碳酸氢铵的结晶母液。
32.优选的,对所述结晶母液进行分解处理,将分解得到的二氧化碳和氨进行回收,将分解余液用于补水液流和/或在步骤(4)中进行利用。
33.本发明有益效果在于:
34.1、本发明所使用的棒状二氧化铈催化剂相比常见的球形、不定型二氧化铈催化剂活性更高,同时,相对现有技术采用的20-50nm氧化铈催化剂,本发明的棒状氧化铈颗粒更大,更容易沉降和从水解液中分离,经实验证明且具备更好的抗污染能力。
35.2、基于目前氧化铈催化面临的长时间接触反应液易污染失活、难以有效分离等问题,本发明工艺优先考虑将二氧化铈催化的步骤仅放置于2-氨基-4-甲硫基丁腈水解为蛋氨酸酰胺的酰胺化反应过程,有效缩短了二氧化铈参与水解反应的时长和工艺流程;另外,氧化铈催化剂不完全滤除蛋氨酸酰胺水解液而获得回收和/或利用,相对于完全固液分离的方式下氧化铈催化剂固体层充当“滤芯”的角色,本发明工艺能够有效避免大量增加反应体系中聚合物、杂质色素等包裹吸附催化剂的情况发生,使昂贵的二氧化铈可循环使用周期更长。
36.3、相对于现有使用减压蒸馏或浓缩方式脱氨获得蛋氨酸,本发明采用对蛋氨酸水解液进行二氧化碳酸化的方式取出蛋氨酸,结晶时间短,蛋氨酸取出率高(一次取晶率可达85.71%),结晶母液可通过分解回收二氧化碳和氨,分解余液进一步回收利用。另外,采用本发明结晶方式,蛋氨酸结晶母液汇总杂质产生和累积的速度更慢,有利于母液健康循环从而使催化剂也能更长久地使用。
具体实施方式
37.所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整所获得的技术方案,仍属于本发明的保护范围。
38.实施例1(氨化,氰醇:氨,1:5)
39.将151.76g氰醇(86.4w%,1mol)转入1l锆材316l高压釜中,加入340g氨水(25w%,5mol),密闭反应釜,开启加热和搅拌,升温至50℃,压力为0.4mpa,保温反应45min,hplc检测反应液中氰醇转化率达99.5%,降温体系至常温,获得2-氨基-4-甲硫基丁腈物料,用于后续催化制备蛋氨酸反应。
40.实施例2
41.将按实施例1方法制备得369.3g 2-氨基-4-甲硫基丁腈物料(35.2w%)与200g补水物流加入316l锆材高压釜中,加入催化剂a二氧化铈172g(棒状二氧化铈,粒径10nm,长100nm,表面积86.3m2/g)(1.0eq),在80℃搅拌1h,过滤除去催化剂a,hplc分析反应液,2-氨基-4-甲硫基丁腈转化为蛋氨酸及其铵盐转化率为99.9%。回用催化剂a重复用于催化2-氨基-4-甲硫基丁腈制备蛋氨酸,分析并记录循环15次的转化率变化见表1。
42.实施例3
43.操作同实施例2,催化剂更换为催化剂b(棒状二氧化铈,粒径30nm,长200nm,表面积45m2/g)。回用催化剂b循环催化15次,分析记录循环过程转化率变化见表1。
44.实施例4
45.操作同实施例2,催化剂更换为催化剂c(球状二氧化铈,粒径30nm,表面积110m2/g)。回用催化剂c循环催化5次,分析记录循环过程转化率变化见表1。
46.实施例5
47.操作同实施例2,催化剂更换为催化剂d(球状二氧化铈,粒径200nm,表面积23m2/g)。回用催化剂d循环催化5次,分析记录循环过程转化率变化见表1。
48.实施例6
49.操作同实施例2,催化剂更换为催化剂e(球状二氧化铈,粒径1μm,表面积10m2/g)。回用催化剂e循环催化5次,分析记录循环过程转化率变化见表1。
50.实施例7
51.操作同实施例2,催化剂更换为催化剂f(方块二氧化铈,粒径30nm,表面积110m2/g)。回用催化剂f循环催化10次,分析记录循环过程转化率变化见表1。
52.实施例8
53.操作同实施例2,催化剂更换为催化剂g(无定型二氧化铈,粒径20nm,表面积139m2/g)。回用催化剂g循环催化10次,分析记录循环过程转化率变化见表1。
54.实施例9
55.操作同实施例2,催化剂更换为催化剂h(类球形、八面体混合二氧化铈,粒径30nm,表面积110m2/g)。回用催化剂h循环催化10次,分析记录循环过程转化率变化见表1。
56.实施例10(一步催化,非完全分离,离交树脂,95℃)
57.将按实施例1方法制备的419.4g2-氨基-4-甲硫基丁腈物料(31w%)与200g补水物流送入316l锆材高压釜中,加入172g催化剂a(棒状二氧化铈,粒径10nm,长100nm,表面积86.3m2/g)(1.0eq),在60℃搅拌30min进行酰胺化反应,hplc检测反应液中2-氨基-4-甲硫基丁腈转化率99.7%,获得蛋氨酸酰胺反应体系静置分层后倾析上层清液得到蛋氨酸酰胺水解液a和下层浸于溶液组分的催化剂体系。蛋氨酸酰胺水解液a进一步经微滤膜过滤得蛋氨酸酰胺水解液b,将上述蛋氨酸酰胺水解液b与酸性磺酸基离交树脂(聚苯乙烯磺酸基)在
95℃接触反应1h,得到蛋氨酸水解液。
58.重复上述反应步骤,并将催化剂体系回用于反应中,循环套用30次,分析结果并记载于表1.
59.实施例11(一步催化,非完全分离,固体超强酸,140℃)
60.将按实施例1方法制备的469.3g2-氨基-4-甲硫基丁腈物料(27.7w%)与220g补水物流送入316l锆材高压釜中,加入172g催化剂b(棒状二氧化铈,粒径30nm,长200nm,表面积45m2/g)(1.0eq),密闭反应釜,在70℃搅拌30min进行酰胺化反应,hplc检测反应液中2-氨基-4-甲硫基丁腈转化率99.5%,获得蛋氨酸酰胺反应体系静置分层后倾析上层清液得到蛋氨酸酰胺水解液517g(蛋氨酸酰胺及蛋氨酸铵含量21.5%),和下层浸于溶液组分的催化剂体系。将上述蛋氨酸酰胺水解液与so
42-/fe2o3固体超强酸在140℃接触反应50min,得到蛋氨酸水解液511g(蛋氨酸含量21.6%),收率98.6%;
61.重复上述反应步骤,并将催化剂体系d回用于反应中,循环套用30次,分析结果并记载于表1。
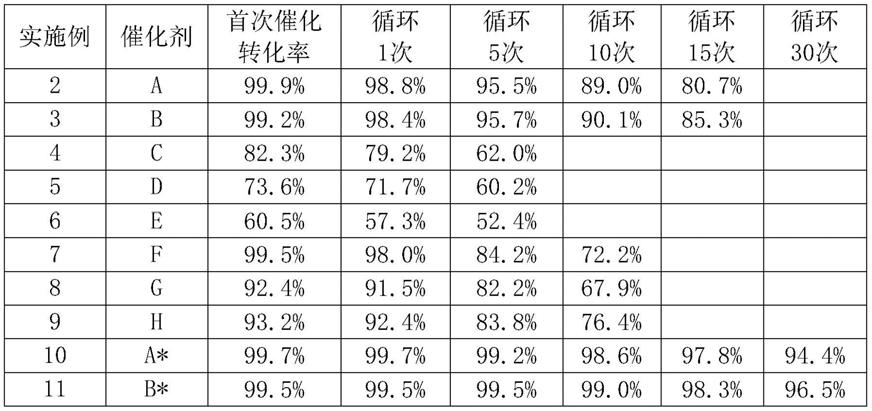
62.表1不同催化剂循环催化2-氨基-4-甲硫基丁腈转化率变化对比
[0063][0064]
注:实施例10、11中二氧化铈催化剂相对实施例2、3催化剂未改变,但与实施例2、3直接将2-氨基-4-甲硫基丁腈水解为蛋氨酸水解液不同,其仅参与2-氨基-4-甲硫基丁腈水解为蛋氨酸酰胺反应体系后即被分离循环。
[0065]
实施例12(一步催化、0.6eq、脱氨、固体酸水解)
[0066]
将按实施例1方法制备的462.6g2-氨基-4-甲硫基丁腈物料(28.1w%)与200g补水物流送入316l锆材高压釜中,加入103.2g催化剂b(棒状二氧化铈,粒径30nm,长200nm,表面积45m2/g)(0.6eq),密闭反应釜,在80℃搅拌反应40min进行酰胺化反应,hplc检测反应液中2-氨基-4-甲硫基丁腈转化率99.5%(2-氨基-4-甲硫基丁腈残留0.04%),获得蛋氨酸酰胺反应体系静置分层后倾析上层清液得到蛋氨酸酰胺水解液496.5g,蒸汽汽提脱除蛋氨酸酰胺水解液中游离氨(游离氨低于0.5%),脱氨后与负载于管式反应器中的so
42-/zro2固体超强酸在80-160℃阶梯升温中接触反应35min,获得蛋氨酸水解液456g(蛋氨酸含量24.5%),将蛋氨酸水解液转入夹套结晶釜,通入二氧化碳酸化结晶60min,ph降至7.26,固液分离获得蛋氨酸固体滤饼,纯水洗涤滤饼2次105℃烘干得蛋氨酸产品95.76g,纯度99.5%,蛋氨酸一次取晶率85.71%。
[0067]
实施例13(减压蒸馏结晶)
[0068]
按实施例12制备得蛋氨酸水解液487g(蛋氨酸含量22.95%),在100℃(-0.5~0.6mpa)减压蒸馏结晶2h,分离得蛋氨酸固体和结晶母液,蛋氨酸固体烘干得72.73g,纯度98.6%,蛋氨酸一次取晶率为65.08%。
[0069]
实施例14(蒸馏 二氧化碳酸化)
[0070]
将按实施例1方法制备的2-氨基-4-甲硫基丁腈物料与补水物流送入316l锆材高压釜中,加入催化剂b(棒状二氧化铈,粒径30nm,长200nm,表面积45m2/g)(0.6eq),密闭反应釜,在80℃搅拌反应40min进行酰胺化反应,hplc检测反应液中2-氨基-4-甲硫基丁腈转化率99.5%(2-氨基-4-甲硫基丁腈残留0.04%),获得蛋氨酸酰胺反应体系静置分层后倾析上层清液得到蛋氨酸酰胺水解液,减压蒸馏脱除蛋氨酸酰胺水解液中游离氨,脱氨后与负载于管式反应器中的固体超强酸在80-160℃阶梯升温中接触反应,获得蛋氨酸水解液,将蛋氨酸水解液于90℃减压蒸馏(-0.04~0.06mpa)脱氨1h,固液分离获得蛋氨酸产品1和一次结晶母液,将一次结晶母液转入夹套结晶釜,通入二氧化碳酸化二次结晶40min,固液分离获得二次结晶蛋氨酸固体滤饼,滤饼与蛋氨酸产品1合并用纯水洗涤滤饼2次,105℃烘干洗涤后蛋氨酸固体得蛋氨酸产品合并139.68g,纯度99.2%,收率93%,洗水与结晶母液合并待用。
[0071]
实施例15(结晶母液分解和回用)
[0072]
将实施例12结晶母液361g(17.5%碳酸氢铵,4.44%蛋氨酸)送入小型分解塔在180℃分解回收二氧化碳和氨,塔底得分解余液334g,分析分解余液中蛋氨酸4.78%,铵根1.13%,碳酸氢铵脱除率73.8%。
[0073]
进一步,可以对分解余液继续进行浓缩、通入二氧化碳酸化再次回收蛋氨酸,也可将其循环回结晶步骤或用作2-氨基-4-甲硫基丁腈补水。
[0074]
实施例16(回用5次,色度)
[0075]
将按实施例1方法制备的2-氨基-4-甲硫基丁腈物料与按实施例15获得的分余解液回用补水物流送入316l锆材高压釜中,加入催化剂k(棒状二氧化铈,粒径50nm,长300nm,表面积32m2/g)(0.8eq),密闭反应釜,在60℃搅拌反应65min进行酰胺化反应,hplc检测反应液中2-氨基-4-甲硫基丁腈转化率99.6%(2-氨基-4-甲硫基丁腈残留0.02%),获得蛋氨酸反应体系静置分层后倾析上层清液得到蛋氨酸酰胺水解液,减压蒸馏脱除蛋氨酸酰胺水解液中游离氨后与负载于管式反应器中的固体超强酸在80-160℃阶梯升温中接触反应,获得蛋氨酸水解液,将蛋氨酸水解液转入夹套结晶釜,通入二氧化碳酸化结晶60min,固液分离获得蛋氨酸固体滤饼和结晶母液,纯水洗涤滤饼2次105℃烘干得蛋氨酸产品,纯度99.2%,结晶母液送入分解塔中分解获得二次分解余液。套用分解液5次,获得五次分解余液,使用加德纳比色管对比色度为8号。
[0076]
实施例17(回用5次,色度杂质)
[0077]
按对实施例13方法制备蛋氨酸并获得结晶母液,将结晶母液返回补水套用,循环5次,获得5次套用结晶母液,使用加德纳比色管对比色度为12号。
[0078]
通过以上实施例12与实施例13可以看出,对蛋氨酸水解液采用不同的结晶方式会影响最后产品的纯度与取晶率,本发明采用的工艺获得的蛋氨酸的纯度与取晶率更高;通过实施例16与实施例17的色度对比可知,本发明生产工艺中杂质产生和积累更慢,更有利
于母液健康循环从而使催化剂也能更持久地使用。
[0079]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。