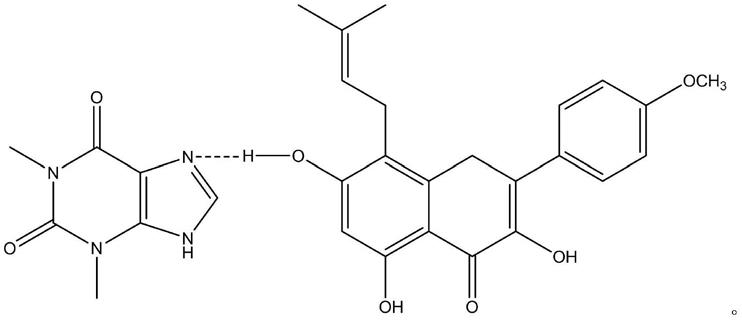
1.本发明涉及药物共晶领域,特别涉及淫羊藿苷元有机药物共晶技术领域,具体涉及一种淫羊藿苷元与茶碱的药物共晶及其制备方法和应用。
背景技术:
2.淫羊藿苷元(icaritin,it)为小檗科淫羊藿属植物淫羊中的一种多羟基黄酮类单体成分。药理研究表明,淫羊藿苷元抗骨质疏松活性较淫羊藿中其他黄酮苷类化合物强,在体外具有促进成骨细胞活性,抑制破骨细胞活性的作用。传统中药淫羊藿具有补肾助阳,强筋健骨,祛风除湿,洗疮杀虫,消疲化痛之功效。淫羊藿苷作为其主要有效成分之一,近年来吸引了国内外众多学者的关注,并对其药理作用进行了深人而广泛的研究,迄今的结果发现淫羊藿苷元的主要生理活性在于改善心脑血管系统功能、增强机体免疫力及调节内分泌,同时还具有抗肿瘤、抗肝毒、抗缺氧再氧合和健骨等作用。
3.虽然淫羊藿苷元具有较好的临床应用前景,但是,其较差的水溶性极大的限制了其临床应用。淫羊藿苷元在水中的溶解度不足1μg/ml,其较差的水溶性导致其口服吸收差,生物利用度低的问题比较突出。目前关于解决淫羊藿苷元的溶解性的研究主要集中在两个方面,一种是研究制备淫羊藿苷元或其衍生物的新晶型,一种比较常规的是通过制剂的手段改善其溶解性。比如贾东升等通过制备淫羊藿苷元磷脂复合物显著改善了原料药与物理混合物的溶解性(贾东升,赵江丽,施峰等.淫羊藿苷元磷脂复合物的制备及固体分散体研究[j].中草药,2010,9(41)),但是该方法存在溶出介质可能破坏药物结构或溶出介质蒸发的问题;王晋艳等通过微乳技术对淫羊藿苷元进行增溶研究(晋艳,陈彦,张振海等.淫羊藿苷元自微乳在caco-2细胞模型的肠吸收特性初步研究[j].中草药,2012,3(43)),虽然能够改善淫羊藿苷元的溶解与吸收,但是其工艺过程中使用了大量的乳化剂和助乳化剂,存在一定的质量安全隐患。
[0004]
而关于对淫羊藿苷元原料形式的研究报道较少,专利cn101200743a虽然报道了制备出了晶体状的淫羊藿苷元,但是并没有改善其溶解特性。专利cn104860958a报道了脱水淫羊藿素的晶型a和晶型b,且研究结果表明较无定形粉末的脱水淫羊藿素其溶解性有一定的改善,但是并未有显著的提高。
[0005]
目前关于淫羊藿苷元的相关报道较多,但是主要是关于其制备、制剂、理化性质及药理等性质的报道,但是并没有显著性的改善淫羊藿苷元的溶解性。因此仍然需要研究新的显著改善淫羊藿苷元溶解性的晶体,可以有效改善其理化性质,从根本上解决淫羊藿苷元的溶解性问题,尽量减少制剂生产过程中辅料的加入,尽可能的减少用药隐患。
技术实现要素:
[0006]
鉴于现有技术报道的淫羊藿苷元溶解性差及现有的改善方法存在弊端的问题,本发明通过共晶技术成功制备获得了一种淫羊藿苷元-茶碱共晶,该共晶体可以显著的改善淫羊藿苷元的溶解特性,可以显著的改善淫羊藿苷元作为制剂活性成分时的溶解性和溶出
特性,并且具有较好的理化稳定性。
[0007]
本发明第一方面,提供了淫羊藿苷元-茶碱共晶,在其晶体结构中淫羊藿苷元与茶碱以如下方式结合:
[0008][0009]
优选地,所述共晶体中,淫羊藿苷元与茶碱的摩尔比为1:1。
[0010]
优选地,所述的淫羊藿苷元-茶碱共晶,使用cu-kα辐射,以2θ表示的x射线衍射谱图在7.3
±
0.2
°
,9.1
±
0.2
°
,16.0
±
0.2
°
,26.1
±
0.2
°
有特征峰。
[0011]
优选地,所述的淫羊藿苷元-茶碱共晶,使用cu-kα辐射,以2θ表示的x射线衍射谱图在7.3
±
0.2
°
,9.1
±
0.2
°
,9.7
±
0.2
°
,14.5
±
0.2
°
,16.0
±
0.2
°
,16.8
±
0.2
°
,26.1
±
0.2
°
,39.8
±
0.2
°
有特征峰。
[0012]
优选地,所述的淫羊藿苷元-茶碱共晶,使用cu-kα辐射,其特征峰符合如图1所示的x射线粉末衍射图谱。
[0013]
优选地,所述的淫羊藿苷元-茶碱共晶,其在差示扫描量热曲线(dsc)中存在一个吸热峰225.59℃。
[0014]
优选地,本发明第二方面,提供一种淫羊藿苷元-茶碱共晶的制备方法,具体制备步骤包括:将淫羊藿苷元和茶碱溶于有机溶剂中,加热搅拌,降温析晶,过滤干燥得淫羊藿苷元-茶碱共晶。
[0015]
优选地,所述的有机溶剂选自乙腈、丙酮、四氢呋喃、甲醇、乙醇、异丙醇中的一种或几种。
[0016]
进一步优选地,所述有机溶剂选自乙腈、丙酮的一种或两种。
[0017]
优选地,所述的淫羊藿苷元与茶碱的摩尔比为1:0.9~1.2;优选为1:1.05~1.1。
[0018]
优选地,所述的淫羊藿苷元和有机溶剂的质量体积比为5~7:1,其中质量以mg计,体积以ml计。
[0019]
优选地,所述的加热搅拌的时间为2~6h;优选为3~4h。
[0020]
优选地,所述的溶解加热的温度为45~75℃。
[0021]
优选地,所述的降温析晶温度为0~15℃;优选为5~10℃。
[0022]
优选地,所述的析晶时间为3~5h。
[0023]
进一步优选地,所述制备方法包括以下步骤:
[0024]
将淫羊藿苷元和茶碱溶于有机溶剂中,45~75℃加热溶解,搅拌回流反应2~6小时,降温至5~10℃析晶3~5h小时,过滤,洗涤滤饼,干燥得淫羊藿苷元茶碱共晶。
[0025]
优选地,所述洗涤滤饼的溶剂选自乙醇、乙腈或丙酮中的一种。
[0026]
优选地,所述干燥温度为50~70℃,干燥时间为6~8小时。
[0027]
本发明第三方面,提供一种药物组合物,该组合物含本发明所述的淫羊藿苷元-茶碱共晶,并混有其它组分。
[0028]
优选地,本发明的药物组合物制备如下:使用标准和常规的技术,使本发明化合物与制剂学上可接受的固体或液体载体结合,以及使之任意地与制剂学上可接受的辅助剂和赋形剂结合制备成药用剂型。
[0029]
优选地,所述的其它组分包括可联合使用的药物活性成分、赋形剂、填充剂等。
[0030]
优选地,所述的药物组合物为喷雾剂、片剂、胶囊剂、粉针剂、注射剂等。
[0031]
本发明第四方面,供一种淫羊藿苷元-茶碱共晶作为活性成分制备治疗疾病药物中的用途;优选地的疾病包括各种炎症、免疫性疾病、肿瘤、高血糖症、贫血及心脑血管疾病等疾病。
[0032]
晶体结构的确认
[0033]
x射线晶体数据在日本理学xtalab synergy型号仪器上收集,测试温度293(2)k,用cuka辐射,以ω扫描方式收集数据并进行lp校正。晶体结构用01ex-2软件中的shelxt程序计算得到,并采用shelxl程序通过最小二乘法修正结构参数和判别原子种类,使用几何计算法和差值fourier法获得全部氢原子位置,拟优合度(goof值)1.019,接近于1.0,表明权重方案合适,结构准确。
[0034]
测试及解析本发明制备的淫羊藿苷元-茶碱共晶所得晶体学数据是(表1):其晶体学参数是:三斜晶系,空间群为p-1;晶胞参数为:1;晶胞参数为:α=97.4527(9)
°
,β=108.6420(10)
°
,γ=114.5138(12)
°
,晶胞体积,晶胞体积分子式是:c
28h28
n4o8,分子量是:548.54。本发明的淫羊藿苷元-茶碱共晶的ortep图表明,淫羊藿苷元和茶碱通过分子内氢键连接在一起,其中淫羊藿苷元羟基h3与茶碱上的n3形成氢键,淫羊藿苷元的羟基上h4与酮基o5形成分子内氢键,该共晶的堆积图如附图4所示。
[0035]
表1淫羊藿苷元-茶碱共晶主要晶体学数据
[0036][0037]
本发明中x-射线粉末衍射测试仪器及测试条件:x-射线粉末衍射仪:panalytical e;cu-kα;样品台:平板;入射光路:bbhd;衍射光路:plxcel;电压45kv,电流40ma;发散狭缝:1/4;防散射狭缝:1;索拉狭缝:0.04rad;步长:0.5s;扫描范围:3~50
°
。
[0038]
依据晶体学数据,其对应的x射线粉末衍射图(cu-kα)中特征峰详见附图1及表2。
[0039]
表2.淫羊藿苷元-茶碱共晶主要的pxrd峰
[0040][0041]
依照本发明的方法在实施例中所制备的所有样品基本符合x射线粉末衍射谱图。
[0042]
本发明中tga/dsc热分析测试仪及测试条件:tga/dsc热分析仪:mettler toledo tga/dsc3 ;动态温度段:30~300℃;加热速率:10℃/min;程序段气体n2;气体流量:50ml/min;坩埚:铝坩埚40μl。
[0043]
本发明所述方法制备的淫羊藿苷元茶碱共晶的tga/dsc测试结果如图2所示,dsc检测结果有一个吸热峰,对应温度为225.59℃。根据tga检测结果可以看出存在一个失重台阶,计算表明该淫羊藿苷元茶碱共晶不存在溶剂合物。
[0044]
本发明提供的制备淫羊藿苷元-茶碱共晶晶体的方法操作简便,制备的晶体纯度高,本发明提供的淫羊藿苷元-茶碱共晶晶体具有较好的稳定性,并且显著的改善了淫羊藿苷元的溶解性,可以显著的改善淫羊藿苷元作为制剂活性成分时的溶解性和溶出特性,为淫羊藿苷元的制剂生产提供了一种优质的原料选择。
附图说明
[0045]
图1:淫羊藿苷元-茶碱共晶的x射线粉末衍射图谱。
[0046]
图2:淫羊藿苷元-茶碱共晶的dsc-tga图。
[0047]
图3:淫羊藿苷元-茶碱共晶的ortep图。
[0048]
图4:淫羊藿苷元-茶碱共晶的堆积图。
具体实施方式
[0049]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属本发明要求保护的范围。
[0050]
实施例1
[0051]
将368mg淫羊藿苷元,189mg茶碱加入到30ml丙酮和40ml乙醇的混合溶剂中,加热到50℃搅拌回流反应3小时,缓慢降温至5~10℃后,控温静置析晶4小时,过滤,滤饼用乙醇洗涤,50℃下真空干燥8h得淫羊藿苷元茶碱共晶,收率90.33%,纯度99.95%。
[0052]
实施例2
[0053]
将368mg淫羊藿苷元,198mg茶碱加入到30ml乙腈和40ml丙酮的混合溶剂中,加热到50℃搅拌回流反应4小时,缓慢降温至5~10℃后,控温静置析晶4小时,过滤,滤饼用乙腈洗涤,50℃下真空干燥6h得淫羊藿苷元茶碱共晶,收率89.68%,纯度99.88%。
[0054]
实施例3
[0055]
将368mg淫羊藿苷元,216mg茶碱加入到30ml四氢呋喃和30ml乙醇的混合溶剂中,加热到60℃搅拌回流反应5小时,缓慢降温至5~10℃后,控温静置析晶4小时,过滤,滤饼用乙醇洗涤,50℃下真空干燥7h得淫羊藿苷元茶碱共晶,收率91.25%,纯度99.32%。
[0056]
实施例4
[0057]
将368mg淫羊藿苷元,189mg茶碱加入到52ml丙酮中,加热到50℃搅拌回流反应5小时,缓慢降温至5~10℃后,控温静置析晶5小时,过滤,使用5℃左右的丙酮洗涤滤饼,50℃下真空干燥7h得淫羊藿苷元茶碱共晶,收率87.56%,纯度99.35%。
[0058]
实施例5
[0059]
将368mg淫羊藿苷元,189mg茶碱加入到70ml乙腈中,加热到50℃搅拌回流反应5小时,缓慢降温至5~10℃后,控温静置析晶4小时,过滤,滤饼用乙腈洗涤,50℃下真空干燥6h得淫羊藿苷元茶碱共晶,收率87.36%,纯度99.21%。
[0060]
稳定性实验
[0061]
具体的稳定性试验方法参照中国药典2015版第四部有关稳定性考察的指导方法进行,纯度检测用hplc法进行检测,具体的检测结果见表3。
[0062]
表3淫羊藿苷元-茶碱共晶在光照、高温及高湿条件下的稳定性试验结果
[0063][0064][0065]
溶解性实验
[0066]
方法:分别量取10ml的介质(水、0.01mol/lhcl溶液)于西林瓶中,加入过量的待测样品,将西林瓶密封置于25℃恒温水浴中搅拌1小时,经0.45μm滤膜过滤,取滤液;在270nm的波长处分别测定吸光度,通过测试标准对照品的吸光度来计算其溶解度。
[0067]
表4淫羊藿苷元-茶碱共晶在不同介质中的溶解度
[0068]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。