优克那非氧化物及其制备方法和用途
1.本技术要求享有2020年11月2日向中国国家知识产权局提交的申请号为202011205349.0,名称为“优克那非氧化物及其制备方法和用途”的在先发明专利申请的优先权权益。该申请的全文通过引用的方式结合至本文。
技术领域
2.本发明涉及药物化学领域,具体涉及优克那非氧化物及其制备方法和用途。
背景技术:
3.勃起功能障碍(erectile dysfunction,ed)是指持续不能达到和/或)维持充分勃起以获得满意的性生活。据统计目前全球范围内约有众多男性患有不同程度ed,到2025年该患病人数将会增加一倍。ed的治疗选择方案较多,其中选择性磷酸二酯酶5(pde5)抑制剂是目前研究最为成熟的ed治疗药物,也是临床治疗ed的一线药物。如今已批准上市的该类药物共有五种,分别为西地那非(sildenafil)、他达拉非(tadalafil)、伐地那非(vardenafil)、乌地那非(udenafil)和米罗那非(mirodenafil)。
4.专利文献cn100374441c公开了一系列吡咯并嘧啶酮结构化合物以及其制备方法和用途。其中化合物1-hcl,即2-[2-乙氧基-5-(4-乙基哌嗪-1-磺酰)苯基]-5-甲基-7-正丙基-3,7-二氢吡咯并[2,3-d]嘧啶-4-酮单盐酸盐已在该文献实施例1中公开,其为盐酸优克那非。
[0005]
专利文献cn104530054b公开了盐酸优克那非多晶型及其制备方法,该专利文献报道的晶型a有良好的物理和化学稳定性,易于工业化生产,为一种药学上可接受的、稳定的盐酸优克那非新晶型。
[0006]
然而,优克那非及其盐的质量控制方法和/或其用药安全性和稳定性的改善仍存在持续的需求和不确定性,其研究结果对于该药物的应用和开发均具有非常重要的意义。
技术实现要素:
[0007]
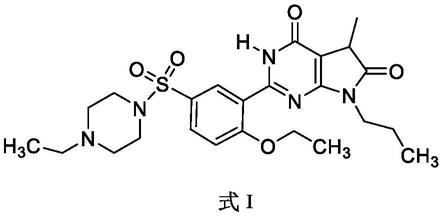
为改善上述技术问题,本发明提供如下式i所示的化合物或其药学上可接受的盐:
[0008][0009]
本发明还提供一种药物组合物,包含优克那非或其药学上可接受的盐,以及药学上可接受的辅料。
[0010]
根据本发明的实施方案,所述药物组合物具有避光和/或密封的包装。优选地,所述药物组合物具有避光和密封的包装。
[0011]
根据本发明的实施方案,所述包装的材料包括内包材,或者包括内包材和外包材。
[0012]
作为实例,所述内包材可以选自聚氯乙烯、聚偏二氯乙烯和铝箔(如药用铝箔)中的一种、两种或三种。
[0013]
作为实例,所述外包材可以选自复合膜,例如包含聚酯、铝和聚乙烯中的两种或三种的复合膜。
[0014]
本发明还提供一种药盒,所述药盒包括盒体和位于所述盒体内的本发明的药物组合物,例如包括盒体和位于所述盒体内的具有避光和/或密封的包装的所述药物组合物。
[0015]
根据本发明的实施方案,所述药物组合物中,以式i所示的化合物计,式i所示的化合物或其药学上可接受的盐的重量百分比含量≤0.5%。
[0016]
根据本发明的实施方案,所述药物组合物中,以式i所示的化合物计,式i所示的化合物或其药学上可接受的盐的重量百分比含量可以≥0。
[0017]
根据本发明的实施方案,所述药物组合物中,以式i所示的化合物计,式i所示的化合物或其药学上可接受的盐的重量百分比含量可以为0.001%、0.01%、0.02%、0.03%、0.04%、0.05%、0.06%、0.07%、0.08%、0.09%、0.10%、0.11%、0.12%、0.13%、0.14%、0.15%、0.16%、0.17%、0.18%、0.19%、0.20%、0.25%、0.30%、0.35%、0.40%、0.45%或0.50%,优选≤0.2%,更优选≤0.15%,更优选≤0.1%。
[0018]
本发明还提供式i所示的化合物或其药学上可接受的盐的制备方法,包括将优克那非或其药学上可接受的盐氧化,得到所述式i所示的化合物或其药学上可接受的盐。
[0019]
根据本发明的实施方案,所述氧化在氧化剂存在下进行。例如,所述氧化剂为过氧化氢、c
1-10
的过氧脂肪酸、过氧苯甲酸、卤代过氧苯甲酸、过硫酸铵、过氧化氢异丙苯和过氧化氢二异丙苯中的至少一种,优选为过氧化氢。
[0020]
根据本发明的实施方案,所述氧化剂与优克那非或其药学上可接受的盐的质量比为0.3:1~1:1,优选为0.4:1~0.6:1,示例性为0.3:1、0.4:1、0.5:1、0.6:1、0.7:1、0.8:1、0.9:1、1:1。
[0021]
根据本发明的实施方案,氧化反应的温度为20~100℃,优选为50~100℃,更优选为75~85℃,示例性为20℃、30℃、40℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃、100℃。
[0022]
根据本发明的实施方案,所述氧化在溶剂中进行。例如,所述溶剂选自所述氧化反应的温度下为液体的有机酸,优选为c
1-10
的有机酸,更优选为甲酸、乙酸、丙酸、丁酸、异丁酸、正戊酸、1-甲基丁酸、2-甲基丁酸、2,2-二甲基丙酸中的一种、两种或更多种,示例性为甲酸。
[0023]
根据本发明的实施方案,可以在加入所述氧化剂前对含有优克那非或其药学上可接受的盐的溶液进行降温处理。例如,降温至温度为-10~10℃,优选为-5~5℃。优选地,所述氧化剂加入含有优克那非或其药学上可接受的盐的溶液中时的温度为-10~10℃,优选为-5~5℃,更优选地,氧化剂与所述溶液的温度相同。
[0024]
根据本发明的实施方案,所述溶剂与优克那非或其药学上可接受的盐的质量比为3:1~15:1,优选为7:1~10:1,示例性为3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1。
[0025]
根据本发明的实施方案,所述制备方法还包括后处理步骤。
[0026]
根据本发明的实施方案,所述后处理步骤包括向氧化体系中加入还原剂,淬灭反
应。
[0027]
根据本发明的实施方案,所述还原剂为亚硫酸钠、亚硫酸氢钠和硫酸亚铁中的一种、两种或更多种,优选为亚硫酸钠和/或亚硫酸氢钠。
[0028]
根据本发明的实施方案,所述还原剂以溶液形式加入氧化体系。优选地,还原剂溶液的浓度为0.1mol/l~5.0mol/l,优选为0.5mol/l~3mol/l,示例性为0.5mol/l、1.0mol/l、1.5mol/l、2mol/l、3mol/l、4mol/l、5mol/l。
[0029]
根据本发明的实施方案,所述制备方法还包括纯化步骤。优选地,所述纯化步骤位于后处理步骤之后。
[0030]
根据本发明的实施方案,所述纯化步骤包括上述后处理完成后,对反应产物进行萃取、洗涤、干燥和重结晶的步骤。
[0031]
根据本发明的实施方案,待后处理完成后,向反应体系中加入水和第一溶剂,搅拌萃取,得到有机相。
[0032]
根据本发明的实施方案,所述第一溶剂为二氯甲烷、乙酸乙酯和甲基叔丁基醚中的一种、两种或更多种,优选为二氯甲烷。
[0033]
根据本发明的实施方案,对所述有机相进行洗涤。例如,使用饱和氯化钠溶液进行洗涤。
[0034]
根据本发明的实施方案,所述干燥为使用干燥剂对洗涤后的有机相进行干燥。
[0035]
根据本发明的实施方案,所述干燥剂可以为无水硫酸钠、无水硫酸镁和分子筛中的一种、两种或更多种,优选为无水硫酸钠和/或无水硫酸镁。
[0036]
根据本发明的实施方案,所述重结晶步骤包括:过滤干燥后的有机相,浓缩得到的滤液,向浓缩物中加入第二溶剂,升温搅拌,而后降温析晶。
[0037]
根据本发明的实施方案,所述浓缩的温度为20~80℃,优选为30~50℃,示例性为20℃、30℃、40℃、50℃、60℃、70℃、80℃。
[0038]
根据本发明的实施方案,所述第二溶剂为甲醇、乙醇、丙酮、乙酸乙酯、二氯甲烷、乙醚、氯仿、甲苯、二甲苯和乙腈中的一种、两种或更多种,优选为乙醇。
[0039]
根据本发明的实施方案,所述第二溶剂与优克那非或其药学上可接受的盐的质量比为3:1~8:1,优选为3:1~6:1,示例性为3:1、4:1、5:1、6:1、7:1、8:1。
[0040]
根据本发明的实施方案,所述重结晶步骤中,升温至的温度为40~80℃,优选为70~80℃,示例性为40℃、50℃、60℃、70℃、80℃。
[0041]
根据本发明的实施方案,所述降温析晶的温度为-10~30℃,优选为-5~5℃,示例性为-10℃、-5℃、0℃、5℃、10℃、20℃、30℃。
[0042]
根据本发明的实施方案,所述重结晶步骤还包括将析晶得到的产物过滤、干燥。优选地,所述干燥的温度为30~80℃,优选为40~50℃,示例性为30℃、40℃、50℃、60℃、70℃、80℃。
[0043]
根据本发明示例性的方案,所述式i所示的化合物或药学上可接受的盐的制备方法,包括如下步骤:
[0044]
将优克那非或其药学上可接受的盐加入至溶剂中,搅拌溶解而后向其中加入氧化剂进行氧化反应,tlc中控至反应完毕;
[0045]
待反应完毕后,向反应体系中加入还原剂溶液,搅拌;再向体系中加入水和第一溶
剂,搅拌萃取;使用饱和氯化钠溶液洗涤萃取得到的有机相,干燥有机相;
[0046]
过滤上述干燥后的有机相,浓缩滤液,向得到的浓缩物中加入第二溶剂,升温搅拌,随后降温析晶,过滤,将得到的滤饼干燥后,得到目标产物。
[0047]
本发明还提供式i所示的化合物或其药学上可接受的盐的制备方法,包括使优克那非或其药学上可接受的盐经受光的辐照,得到所述式i所示的化合物或其药学上可接受的盐。
[0048]
根据本发明的实施方案,所述的光可以为强白光和/或紫外光,其优选的照度可以为4500lx
±
500lx。
[0049]
本发明还提供式i所示的化合物或其药学上可接受的盐作为标准品或对照品的用途,特别是作为药品标准品或对照品的用途。
[0050]
本发明还提供上述式i所示的化合物或其药学上可接受的盐作为优克那非或其药学上可接受的盐的质量研究标准品或对照品的用途。
[0051]
本发明还提供优克那非或其药学上可接受的盐在制备药物组合物中的用途,其中所述药物组合物具有上文所述的定义。
[0052]
本发明还提供一种治疗和/或预防勃起功能障碍的方法,包括将所述药物组合物施用于有需要的主体。
[0053]
根据本发明的实施方案,所述有需要的主体包括但不限于患有勃起功能障碍的患者或存在勃起功能障碍风险的潜在患者。优选地,所述患者为雄性哺乳动物,特别是男性人类。
[0054]
本发明还提供一种优克那非或其药学上可接受的盐的质量控制方法,包括将式i所示的化合物或其药学上可接受的盐作为标准品或对照品,特别是作为药品标准品或对照品使用。
[0055]
本发明还提供一种优克那非或其药学上可接受的盐的质量控制方法,包括检测式i所示的化合物或其药学上可接受的盐。优选地,所述检测可以是定性检测或定量检测。
[0056]
本发明还提供一种优克那非或其药学上可接受的盐的质量控制方法,包括将优克那非或其药学上可接受的盐在不利于式i所示的化合物或其药学上可接受的盐形成的条件下保存或使用。
[0057]
本发明还提供一种药物组合物的制备方法,包括将优克那非或其药学上可接受的盐与药学上可接受的辅料混合,其中所述制备方法在不利于式i所示的化合物或其药学上可接受的盐形成的条件下进行。
[0058]
本发明还提供一种优克那非或其药学上可接受的盐的保存或使用方法,其中所述保存或使用方法在不利于式i所示的化合物或其药学上可接受的盐形成的条件下进行。
[0059]
优选地,所述“不利于式i所示的化合物或其药学上可接受的盐形成的条件”包括但不限于选自避光和密封条件中的至少一种;更优选选自避光且密封的条件。
[0060]
根据本发明可选的实施方案,所述优克那非或其药学上可接受的盐的保存或使用方法可以在避光和密封,并且在温度低于60℃(优选低于50℃)和/或相对湿度低于rh75%(优选低于65%)的条件下进行。
[0061]
本发明还提供一种式i所示的化合物或药学上可接受的盐的检测方法,包括采用高效液相色谱法检测供试品溶液中式i所示的化合物或药学上可接受的盐。
[0062]
根据本发明的实施方案,所述供试品为含有优克那非或其药学上可接受的盐的药物组合物,优选制剂,例如固体制剂或液体制剂。更优选地,所述药物组合物或制剂包含式i所示的化合物或药学上可接受的盐。
[0063]
根据本发明的实施方案,高效液相色谱法所用的色谱柱为以十八烷基键合硅胶为填充剂的色谱柱;例如,所述色谱柱选自agilent eclipse xdb-c18 250
×
4.6mm,5μm或与其组成和性能相当或接近的色谱柱。
[0064]
根据本发明的实施方案,所述检测的波长为220~230nm,例如222~228nm,优选为226nm。
[0065]
根据本发明的实施方案,色谱柱的柱温为20~30℃,例如22~28℃,示例性为25℃。
[0066]
根据本发明的实施方案,流速为0.5~1.5ml/min,例如0.8~1.2ml/min,示例性为1ml/min。
[0067]
根据本发明的实施方案,所述高效液相色谱法以枸橼酸缓冲液-乙腈作为流动相。优选地,所述流动相中枸橼酸缓冲液的浓度为0.01~0.04mol/l,例如为0.02~0.03mol/l,示例性为0.02mol/l。
[0068]
根据本发明的实施方案,所述枸橼酸缓冲液的ph为5.3~5.5,例如为5.4。
[0069]
根据本发明的实施方案,所述枸橼酸缓冲液为枸橼酸-三乙胺缓冲液。
[0070]
根据本发明的实施方案,所述枸橼酸缓冲液通过将枸橼酸、三乙胺和水混合制备,例如将枸橼酸溶解于水中,用三乙胺调节ph至上述范围。
[0071]
根据本发明的实施方案,所述流动相中枸橼酸缓冲液与乙腈的体积比为(45-55):(55-45),例如为50:50。
[0072]
根据本发明的实施方案,所述供试品溶液由所述流动相配制,所述供试品溶液中,以盐酸优克那非计的浓度为0.5-1.5mg/ml,例如为1.0mg/ml。
[0073]
根据本发明的实施方案,所述检测方法所用的对照品为式i所示的化合物或药学上可接受的盐。
[0074]
根据本发明的实施方案,在所述供试品中,优克那非或其药学上可接受的盐可以以其无定形或晶体形式存在。例如,对于优克那非的盐酸盐(如盐酸优克那非),其可以为多晶型物的形式,例如专利文献cn104530054b中公开的a型多晶型物或b型多晶型物,特别是图1的xrd谱图所示的盐酸优克那非a型多晶型物或图3的xrd谱图所示的盐酸优克那非b型多晶型物。
[0075]
根据本发明的实施方案,式i化合物或其药学上可接受的盐可以以其无定形或晶体形式存在。
[0076]
根据本发明的实施方案,优克那非和式i所示的化合物可以独立地以各种药学上可接受的酸加成盐的形式存在。所述酸加成盐包括但不限于与选自下列的酸的一种形成的盐:盐酸盐、氢氟酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、焦硫酸盐、磷酸盐、硝酸盐、甲磺酸盐、乙磺酸盐、2-羟基乙磺酸盐、苯磺酸盐、甲苯磺酸盐、氨基磺酸盐、2-萘磺酸盐、甲酸盐、乙酰乙酸、丙酮酸、月硅酸酯、肉桂酸酯、苯甲酸盐、醋酸盐、二羟乙酸盐、三氟乙酸盐、三甲基乙酸盐、丙酸盐、丁酸盐、己酸盐、庚酸盐、十一酸盐、硬脂酸盐、抗坏血酸盐、樟脑酸盐、樟脑磺酸盐、柠檬酸盐、富马酸盐、苹果酸盐、马来酸盐、羟基马来酸盐、草酸盐、水杨酸盐、琥珀酸
盐、葡萄糖酸盐、奎尼酸盐、双羟萘酸盐、甘醇酸盐、酒石酸盐、乳酸盐、2-(4-羟基苯甲酰基)苯甲酸盐、环戊烷丙酸盐、二葡糖酸盐、3-羟基-2-萘甲酸盐、烟酸盐、扑酸盐、果胶酯酸盐、3-苯基丙酸盐、苦味酸盐、特戊酸盐、衣康酸盐、三氟甲磺酸盐、十二烷基硫酸盐、对甲苯磺酸盐、萘二磺酸盐、丙二酸盐、己二酸盐、藻酸盐、扁桃酸盐、葡庚酸盐、甘油磷酸盐、磺基水杨酸盐、半硫酸或硫氰酸盐、天冬氨酸盐等。
[0077]
本发明上下文中可将式i化合物称为“优克那非氧化物”。
[0078]
除非另有说明,本发明上下文中的“优克那非”、“优克那非游离碱”、“盐酸优克那非游离碱”意指相同的化合物,即未与酸形成加成盐的优克那非,其化学结构是本领域技术人员已知的,例如实施例中的式ii所示的结构。
[0079]
根据本发明优选的实施方案,式i所示的化合物的药学上可接受的盐选自其盐酸盐。除非另有说明,所述盐酸盐在本发明的上下文中可称为“盐酸优克那非氧化物”、“优克那非氧化物的盐酸盐”或“式i所示化合物的盐酸盐”,这些术语应当被理解为相同的盐型。
[0080]
有益效果
[0081]
本发明通过对优克那非及其药学上可接受的盐的原料药和制剂进行深入研究,确认了式i所示的化合物或其药学上可接受的盐的结构及其制备方法和检测方法,为盐酸优克那非的定性及定量分析提供标准品和对照品,为盐酸优克那非的安全用药提供指导。发明人意外地发现,式i所示的化合物或其药学上可接受的盐会影响优克那非制剂的安全性、有效性和/或稳定性,尤其影响作为药物活性成分的优克那非或其药学上可接受的盐的含量稳定性。因此,控制式i所示的化合物或其药学上可接受的盐有助于改善优克那非或其药学上可接受的盐及其药物组合物的质量,包括其安全性、有效性和/或稳定性。而且,式i所示的化合物或其药学上可接受的盐可作为标准品或对照品,用于优克那非或盐酸优克那非的原料药及其制剂的质量控制。
[0082]
本发明还提供了式i所示的化合物或其药学上可接受的盐的制备方法,原料易得,操作简便,反应条件温和,重复性良好,后处理步骤便捷,精制步骤简洁。
[0083]
本发明还提供一种式i所示的化合物或其药学上可接受的盐的检测方法,可以高精密度和准确度地检测其在原料药和药品中的含量,有利于药品安全性、有效性和/或稳定性的显著改善。
附图说明
[0084]
图1为式i化合物的核磁共振氢谱;
[0085]
图2为式i化合物的核磁共振碳谱;
[0086]
图3为图2中式i化合物的核磁共振碳谱局部放大图;
[0087]
图4为优克那非的核磁共振氢谱;
[0088]
图5为式i化合物的质谱;
[0089]
图6为式i化合物的hplc谱图;
[0090]
图7为盐酸优克那非成品的hplc谱图;
[0091]
图8为加速稳定性实验中第6个月样品的hplc谱图。
具体实施方式
[0092]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0093]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0094]
实施例1
[0095][0096]
向100ml甲酸中加入10g盐酸优克那非的游离碱(即式ii所示的化合物,又称为优克那非),搅拌溶解,降温至-5~5℃,缓慢加入4.5g浓度30%的过氧化氢溶液,加入完毕后缓慢升温至70~80℃,保温反应5h,tlc中控(展开剂:乙酸乙酯与乙醇的体积比=5:1)至反应完毕。向反应液中加入100ml 1mol/l亚硫酸氢钠溶液,搅拌30min,加入纯化水40ml和二氯甲烷200ml,搅拌30min,分液萃取,得到的有机相用饱和氯化钠溶液100ml洗涤后,再用无水硫酸钠干燥2h。过滤,滤液于30~40℃减压浓缩至干,而后加入乙醇40g,升温至70~80℃,体系溶清后降温至-5~5℃,析晶,过滤,滤饼在40~50℃下真空干燥,得到式i所示结构的化合物7.8g,收率75.5%,纯度99.1%。
[0097]
实施例2
[0098][0099]
向45ml甲酸中加入5g式ii所示结构的化合物,搅拌溶解,降温至-5~5℃,缓慢加入3.0g浓度30%的过氧化氢溶液,加入完毕后升温至70~80℃,保温反应4h,tlc中控(展开剂:乙酸乙酯与乙醇的体积比=5:1)至反应完毕。向反应液中加入50ml 1mol/l亚硫酸氢钠溶液,搅拌30min,加入纯化水25ml和二氯甲烷100ml,搅拌30min,分液萃取,得到的有机相用饱和氯化钠溶液50ml洗涤后,再用无水硫酸钠干燥2h。过滤,滤液30~40℃减压浓缩至干,加入乙醇25g,升温至70~80℃,体系溶清后降温至-5~5℃,析晶,过滤,滤饼在40~50℃下真空干燥,得到式i所示结构的化合物3.7g,收率71.6%,纯度99.3%。
[0100]
实施例3
[0101][0102]
向45ml乙酸中加入5g式ii所示结构的化合物,搅拌溶解,降温至-5~5℃,缓慢加入3.0g浓度30%的过氧化氢溶液,加入完毕后升温至70~80℃,保温反应5h,tlc中控(展开剂:乙酸乙酯与乙醇的体积比=5:1)至反应完毕。向反应液中加入50ml 1mol/l亚硫酸氢钠溶液,搅拌30min,加入纯化水25ml和二氯甲烷100ml,搅拌30min,分液萃取,得到的有机相用饱和氯化钠溶液50ml洗涤后,再用无水硫酸钠干燥2h。过滤,滤液30~40℃减压浓缩至干,加入乙醇25g,升温至70~80℃,体系溶清后降温至-5~5℃,析晶,过滤,滤饼在40~50℃下真空干燥,得到式i所示结构化合物3.5g,收率67.8%,纯度96.7%。
[0103]
实施例4
[0104]
利用核磁共振和质谱对实施例1制备得到的产物结构进行确证,测试结果如下:
[0105]
核磁共振谱(bruker av-500型核磁共振仪,dmso-d6)
[0106]
如图1所示,1h-nmrδ(ppm):12.41(h,s),7.96(1h,s),7.87(1h,d,j=8.7hz),7.42(1h,d,j=8.7hz)4.26(2h,q,j=6.8hz),3.58(3h,m),2.92(4h,s),2.42(4h,s),2.31(2h,q,j=7.1hz),1.64(2h,m),1.38(6h,t.j=6.8hz),0.94(3h,t,j=7.0hz),0.86(3h,t,j=7.3hz)。
[0107]
如图2和图3所示,
13
c-nmrδ(ppm):178.23,161.93,159.80,157.33,156.99,132.19,130.01,126.35,122.34,113.47,102.41,65.03,51.12,50.95,45.82,40.23,38.27,21.03,14.10,13.31,11.71,10.98。
[0108]
质谱(agilent 1260-6100lc-ms质谱仪)
[0109]
质谱测试结果如图5所示,504.2[m h]
,502.2[m-h]
。质谱结果与结构理论分子量相符;分子量为奇数,与式i化合物分子中含有奇数个n相符。
[0110]
由上述测试证明,实施例1制备得到的产物为式i所示化合物,即优克那非氧化物。
[0111]
实施例5
[0112]
盐酸优克那非成品中盐酸优克那非氧化物的检测方法:
[0113]
采用实施例1制备的化合物作为对照品,以盐酸优克那非成品(纯度99.8%,单杂0.07%,其核磁共振氢谱如图4所示)作为供试品,采用高效液相色谱(hplc)法对成品中的盐酸优克那非氧化物的含量进行检测。
[0114]
色谱条件:
[0115]
色谱柱:以十八烷基键合硅胶为填充剂(agilent eclipse xdb-c18 250
×
4.6mm,5μm)
[0116]
检测波长:226nm
[0117]
柱温:25℃
[0118]
流速:1ml/min
[0119]
流动相:0.02mol/l枸橼酸缓冲液-乙腈=50:50(体积比);
[0120]
枸橼酸缓冲液的制备过程:取枸橼酸4.2g,加水溶解并稀释至1000ml,用三乙胺调节ph至5.4
±
0.1。
[0121]
供试品溶液:取盐酸优克那非成品适量,精密称定,加流动相溶解并稀释制成每1ml约含盐酸优克那非1.0mg的溶液,摇匀,作为供试品溶液。
[0122]
对照品溶液:取实施例1制备的优克那非氧化物,精密称定,加流动相溶解并稀释制成每1ml约含优克那非氧化物0.01mg的溶液,摇匀,作为对照品溶液。
[0123]
检测结果如图7所示,与对照品的hplc谱图(图6)相对比,供试品溶液中相对保留时间约26.8min的物质即为式i所示结构的优克那非氧化物。
[0124]
上述盐酸优克那非成品中有关物质的检测方法经系统的方法学验证,充分证明了该方法对优克那非氧化物的检出能力。将优克那非氧化物定入盐酸优克那非成品质量标准,并进行严格控制,能够保证盐酸优克那非成品的质量。实施例6盐酸优克那非制剂强降解试验
[0125]
考察的破坏条件:
[0126]
(1)光照:取本品10片,于4500lx
±
500lx强白光和紫外光混合光照条件下放置10天;
[0127]
(2)高温:取本品10片,于60℃条件下避光放置10天;
[0128]
(3)高温 高湿:取本品10片,于60℃
±
2℃,rh75%
±
5%条件下避光放置10天;
[0129]
(4)氧化:取本品10片,加入3%h2o
2 5ml于室温条件下避光放置5h。
[0130]
根据实施例5的方法进行检测,检测结果如下:
[0131]
表1强降解试验考察结果
[0132][0133]
上述结果表明,在高温或高温和高湿条件下,优克那非氧化物有一定程度增长;在光照和氧化条件下,优克那非氧化物增长更加显著。以上结果说明式i化合物为盐酸优克那非制剂的降解产物,为保证盐酸优克那非制剂的质量,需要将其含量控制在本发明所述的范围内。
[0134]
实施例7对herg钾离子通道的抑制作用
[0135]
利用膜片钳技术检测了优克那非和优克那非氧化物对herg通道的阻断作用浓度效应关系,从而评价对心脏herg钾通道抑制作用的风险。
[0136]
细胞培养:
[0137]
采用了稳定表达herg钾通道的hek-293细胞系,herg钾通道细胞购于creacell公司。herg钾通道稳定表达的hek293细胞系在含有10%胎牛血清及0.8mg/ml g418的dmem培养基中培养,培养温度为37℃,二氧化碳浓度为5%。
[0138]
细胞传代:
[0139]
除去旧培养基并用pbs洗一次,然后加入1ml tryple
tm express溶液,37℃孵育0.5min左右。当细胞从皿底脱离,加入约5ml 37℃预热的完全培养基。将细胞悬液用吸管轻
轻吹打使聚集的细胞分离。将细胞悬液转移至无菌的离心管中,1000rpm离心5min收集细胞。扩增或维持培养,将细胞接种于6cm细胞培养皿,每个细胞培养皿接种细胞量为2.5
×
105cells(最终体积:5ml)。为维持细胞的电生理活性,细胞密度必须不能超过80%。
[0140]
试验之前细胞用tryple
tm express分离,将4
×
103细胞铺到盖玻片上,在24孔板中培养(最终体积:500μl),18个小时后,进行试验检测。
[0141]
电生理记录
[0142]
细胞外液:k-007-1
[0143]
140mm nacl,3.5mm kcl,1mm mgcl2·
6h2o,2mm cacl2·
2h2o,10mm d-glucose,10mm hepes,1.25mm nah2po4·
2h2o,naoh调节ph=7.4。
[0144]
细胞内液:k-002-2
[0145]
20mm kcl,115mm k-aspartic,1mm mgcl2·
6h2o,5mm egta,10mm hepes,2mm na
2-atp,koh调节ph=7.2。
[0146]
细胞外液保存时间为2周,细胞内液配好后分装为每管1ml,冻存于-20℃冰箱,每天试验使用新融化的细胞内液。所有细胞内液在三个月内用完。超过三个月,丢弃旧细胞内液,并重新配制。
[0147]
膜片钳检测
[0148]
全细胞膜片钳记录全细胞herg钾电流的电压刺激方案如下:当形成全细胞封接后细胞膜电压钳制于-80mv。钳制电压由-80mv除极至-50mv维持0.5s(作为漏电流检测),然后阶跃至30mv维持2.5s,再迅速恢复至-50mv维持4s可以激发出herg通道的尾电流。每隔10s重复采集数据,观察药物对herg尾电流的作用。以0.5s的-50mv刺激作为漏电流检测。试验数据由epc-10放大器(heka)进行采集并储存于patchmaster(heka)软件中。
[0149]
用微电极拉制仪将毛细玻璃管拉制成记录电极。在倒置显微镜下操纵微电极操纵仪将记录电极接触到细胞上,给予负压抽吸,形成gω封接。形成gω封接后进行快速电容补偿,然后继续给予负压,吸破细胞膜,形成全细胞记录模式。然后进行慢速电容的补偿并记录膜电容及串联电阻。不给予漏电补偿。
[0150]
当全细胞记录的herg电流稳定后开始给药,每个药物浓度作用至5min(或者电流至稳定)后检测下一个浓度,每一个测试化合物检测多个浓度。将铺有细胞的盖玻片置于倒置显微中的记录浴槽中,测试化合物以及不含化合物的外液利用重力灌流的方法从低浓度到高浓度依次流经记录浴槽从而作用于细胞,在记录中利用真空泵进行液体交换。每一个细胞在不含化合物的外液中检测到的电流作为自己的对照组。独立重复检测多个细胞。所有电生理试验在室温下进行。
[0151]
试验结果
[0152]
在三次独立重复试验中检测优克那非和优克那非氧化物对herg通道抑制作用,并通过拟合计算出样品对herg电流的半抑制浓度(ic50),试验结果如下:
[0153]
供试品(优克那非)对herg电流的抑制比例及ic
50
结果:
[0154][0155]
供试品优克那非氧化物对herg电流的抑制比例及ic
50
结果:
[0156][0157]
利用膜片钳技术检测供试品(盐酸优克那非和优克那非氧化物)对herg通道的阻断作用浓度效应关系,从而评价供试品对心脏herg钾通道抑制作用的风险。试验结果表明:在测试浓度范围内,供试品优克那非作用于herg电流的ic50结果为3.9μm,而优克那非氧化物对心脏herg钾通道抑制作用显著强于优克那非,提示优克那非氧化物对心脏herg钾通道抑制作用的风险,故需对所述氧化物杂质的含量进行严格的控制。
[0158]
实施例8盐酸优克那非制剂稳定性试验
[0159]
本实施例中使用的制剂与实施例6中的样品组成相同。
[0160]
本实施例选用的包装材料包括内包材和外包材。其中,内包材选用聚氯乙烯/聚偏二氯乙烯固体药用复合硬片和药用铝箔;外包材选用聚酯/铝/聚乙烯药用复合膜袋。
[0161]
考察条件:在加速条件(40℃
±
2℃,75%
±
5%rh)进行制剂稳定性试验,分别在0月、1月、3月、6月取样检测,结果如下表2所示。
[0162]
表2加速稳定性留样结果
[0163][0164]
根据实施例5的方法,对第6个月的样品进行hplc检测的谱图如图8所示,其中26.782min处为优克那非氧化物的峰。
[0165]
结果表明,在模拟包装材料,加速条件(40℃
±
2℃,75%
±
5%rh)下考察6个月,有关物质中优克那非氧化物缓慢增长,总杂略有增长,其他检测项目均无明显变化,说明该氧化物在上述包装材料条件下得到有效控制。
[0166]
以上对本发明示例性的实施方式进行了说明。但是,本发明的保护范围不拘囿于上述实施方式。凡在本发明的精神和原则之内所作出的任何修改、等同替换或改进等,均应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。