一类glp-1/胃泌素受体双重激动剂及其应用
技术领域
1.本发明涉有生物医药,具体涉及一类glp-1/胃泌素受体双重激动剂及其应用。
背景技术:
2.糖尿病是以高血糖为特征的代谢型疾病,是全世界最主要的慢性非传染病之一。糖尿病可分为胰岛素依赖型糖尿病(1型糖尿病,t1dm)和非胰岛素依赖型糖尿病(2型糖尿病,t2dm),其中t2dm患者占糖尿病患者人数的85%以上。目前治疗t2dm最有效的方法是注射胰岛素,但是在治疗过程中会出现低血糖的危险。受到剂量大小、个体差异、注射途径、注射部位或注射后未进食等因素的影响,胰岛素使用过程可能会出现严重的低血糖反应。因此,寻找安全有效的降血糖新药成为当前糖尿病治疗药物研究的当务之急。
3.glp-1是由末端空肠、回肠和结肠的l细胞所分泌的一种葡萄糖依赖性降血糖多肽激素,与glp-1受体特异性结合后发挥降糖作用。glp-1的主要优点是具有血糖依赖性的肠促胰岛素分泌作用,避免了糖尿病治疗中常存在的产生低血糖症的危险。除了调节血糖,glp-1也可以阻止胰腺β细胞退化,刺激β细胞的增殖和分化,能从源头上改善糖尿病进程。此外,glp-1还具有抑制胃酸分泌、延迟胃排空、抑制食欲等作用,具有部分减重效果。然而,天然glp-1在体内的半衰期很短,这在试图将glp-1作为一种药物的尝试中构成了一个重大的药理学挑战。glp-1在体内会被二肽基肽酶iv(dpp-iv)迅速降解,导致其半衰期仅有几分钟。
4.两栖动物体内的glp-1作用效果与人glp-1类似,所以针对两栖动物glp-1进行结构修饰,有望发现具有更高效和长效降糖作用的新型glp-1类药物。xenglp-1是从非洲爪蟾体内发现的一类动物源属的glp-1类似物,与天然glp-1相比,xenglp-1的降糖活性和稳定性更优。此外,与glp-1相比,除了更为耐受dpp-iv的降解,xenglp-1还显示出对于中性内肽酶(nep)的降解稳定得多。xenglp-1是glp-1受体的高效激动剂,具有许多用天然glp-1观察到的葡萄糖调控作用,许多临床前研究都显示xenglp-1具有若干有益的抗糖尿病特性,包括血糖依赖性的胰岛素合成和分泌增强、胃排空放慢、食物摄入和体重减少,以及促进β细胞增殖和恢复胰岛功能等(biochem.pharmacol.,2017,142,155
–
167;faseb j.,2019,33,7113-7125)。这些效果不仅对于糖尿病人是有益的,并且对罹患肥胖症的患者也是有益的。
5.胃泌素(gastrin)是由胃粘膜细胞和十二指肠g细胞分泌的,它在人体的主要生理作用是刺激胃酸分泌和帮助胃运动。胃泌素的其他作用包括刺激细胞生长,有迹象表明胃泌素可能在胰岛新生中起作用,即刺激胰岛中分泌胰岛素的β细胞生长(见korc,m.,j.clin.invest.,1993,92,1113-1114;rooman et al.diabetes,2002,51,686-690),因此有助于调节血糖。胃泌素和另一种胃肠道激素胆囊收缩素(cholecystokinin,cck)共享受体,它们的受体分为两类,cck-1受体和cck-2受体(gastrin受体),这两种受体对gastrin和cck类似物具有不同的亲和力。cck-1受体主要作为硫酸化cck的受体,而cck-2受体对cck和gastrin具有类似的亲和力。其中cck-2受体也被认为是gastrin受体,因为血浆中gastrin水平高于cck(foucaud et al.reg.peptides,2008,145,17-23)。
6.cck-2受体与配体结合时可启动多种细胞内通路,cck-2受体下游的一个关键途径是mapk(丝裂原活化蛋白激酶)或erk(胞外调节激酶)通路,这些通路也被几种生长激素激活,这是gastrin在细胞增殖作用中的一个关键特征。由于cck-2受体在胰腺中表达,gastrin能够促进胰腺组织的细胞增殖和胰岛再生。gastrin在人体内主要以三种形式存在,按氨基酸数目可分为gastrin-34、gastrin-17和gastrin-14,此外,还存在一种短肽形式,即gastrin-6。gastrin-6的6个氨基酸是gastrin与cck-2受体结合的关键氨基酸,并且其c端是酰胺化形式。
7.wo2005/072045公开了一种“glp-1受体激动剂”和“gastrin化合物”的组合,在预防和/或治疗“glp-1受体激动剂”或“gastrin化合物”已被证明具有治疗效果的条件和/或疾病中具有有益效果。wo2007/095737公开了“exendin-4”和“gastrin化合物”的类似组合,在预防和/或治疗“exendin-4激动剂”或“gastrin化合物”已被证明具有治疗效果的条件和/或疾病中同样具有有益效果。应注意,wo2005/072045或wo2007/095737均未提供任何体内、体外或其他数据来证实其中分别描述和使用的“glp-1受体激动剂”/“gastrin化合物”或“exendin-4”/“gastrin化合物”组合可能有益于例如类型的治疗t2dm。us10406207b2公开了一种exendin-4的截短形式和gastrin-6的缀合肽(zp3022),k.fosgerau等揭示了zp3022在糖尿病模型小鼠中展现出了提高的治疗活性,由于没有长效化修饰,这种缀合肽的半衰期较短,必须频繁注射(diabetes obes metab.,2013,15,62-71)。xinyu chen等揭示了一类xenglp-1与gastrin-6的缀合肽,但是,该类缀合肽如果采用一周一次给药的修饰手段,活性会显著下降,因而只能实现一天一次给药,无法实现一周一次给药(j.med.chem.2020,63,12595-12613)。
技术实现要素:
8.本发明的目的是提供一类新的具有glp-1/胃泌素受体双重激动作用的多肽化合物,所述多肽是基于xenglp-1序列设计的变体,可以同时激动glp-1受体和胃泌素受体,进一步提高了xenglp-1的糖尿病的治疗作用。该类多肽化合物性质稳定,免疫原性低,可以实现一周一次给药。同时多肽化合物具有优异的减重、调脂和降低肝脏脂质含量作用,比单一受体激动剂和已报道glp-1/胃泌素受体双重激动剂在制备用于治疗代谢综合征,诸如糖尿病、肥胖、nafld、nash等疾病的药物方面更具潜力。
9.为实现上述发明目的,本发明的技术方案具体如下:
10.一类glp-1/胃泌素受体双重激动多肽化合物,所述多肽化合物的氨基酸序列通式为:his-aib-glu-gly-thr-tyr-thr-asn-asp-val-thr-glu-tyr-leu-glu-glu-glu-ala-ala-xaa
1-glu-phe-ile-glu-trp-leu-ile-lys-aeea-aeea-asp-styr-nle-glu-trp-nle-asp-phe-nh211.其中:
12.xaa1取自lys或侧链被修饰的lys;
13.其中,侧链被修饰的lys是lys(aeea-aeea-γ-glu-co-(ch2)
n-cooh),结构式如下式所示:
[0014][0015]
其中,n为自然数,且16≤n≤20。
[0016]
优选的,所述n是16、18或20。
[0017]
优选的,所述多肽化合物的氨基酸序列是下列序列之一:
[0018]
(1)seq id no:1
[0019][0020]
(2)seq id no:2
[0021][0022]
本发明还提供了一类glp-1/胃泌素受体双重激动多肽化合物的药学上可接受的盐。
[0023]
优选的,所述盐为glp-1/胃泌素受体双重激动多肽化合物与下述化合物中的一种所形成的盐:氢溴酸、盐酸、甲磺酸、磷酸、乙磺酸、甲酸、对甲苯磺酸、乙酸、乙酰乙酸、丙酮酸、果胶酯酸、丁酸、己酸、苯磺酸、庚酸、十一烷酸、苯甲酸、水杨酸、月桂酸、2-(4-羟基苯甲酰基)苯甲酸、肉桂酸、樟脑酸、环戊烷丙酸、3-羟基-2-萘甲酸、樟脑磺酸、二葡糖酸、烟酸、扑酸、丙酸、过硫酸、、苦味酸、3-苯基丙酸、特戊酸、衣康酸、2-羟基乙磺酸、氨基磺酸、十二烷基硫酸、三氟甲磺酸、萘二磺酸、2-萘磺酸、柠檬酸、扁桃酸、抗坏血酸、酒硬脂酸、石酸、草酸、乳酸、琥珀酸、丙二酸、半硫酸、苹果酸、马来酸、藻酸、富马酸、d-葡糖酸、甘油磷酸、葡庚酸、天冬氨酸、硫氰酸或者磺基水杨酸。
[0024]
本发明还提供了glp-1/胃泌素受体双重激动多肽化合物的药物组合物,该药物组合物包括:以上述任一glp-1/胃泌素受体双重激动多肽化合物或其药学上可接受的盐为有效原料,再加上药学上可接受的载体或稀释剂组成。
[0025]
本发明还提供了含有上述glp-1/胃泌素受体双重激动多肽化合物的药剂,所述的药剂是任何一种药剂学上所说的胶囊、片剂、喷雾剂、吸入剂、注射剂、贴剂、乳剂、膜剂、散剂或者复方制剂,药剂由glp-1/胃泌素受体双重激动多肽化合物和药学上可接受的药用辅料、载体或稀释剂组成。
[0026]
本发明还提供了本发明所述的glp-1/胃泌素受体双重激动多肽化合物、其药学上可接受的盐、其药物组合物或其药剂在制备用于治疗代谢性疾病或病症的药物中的应用。在特定方面,代谢性疾病或病症为糖尿病、nafld、nash或肥胖。在特定方面,糖尿病为1型糖尿病、t2dm。在特定方面,所述药物用于治疗超过一种代谢疾病或病症,例如,糖尿病和nafld;糖尿病和nash;糖尿病和肥胖;肥胖和nash;肥胖和nafld;肥胖合并糖尿病和nafld;或肥胖合并糖尿病和nash。
[0027]
与现有技术相比,本发明的有益效果:
[0028]
与现有的glp-1受体激动剂相比,本发明的glp-1/胃泌素受体双重激动多肽化合
物在更为有效的降低血糖的同时具有更优异的促进胰腺组织的细胞增殖和胰岛再生作用,可以从根本上治疗糖尿病,逆转胰岛素抵抗,治疗肥胖和高血脂,与现有药物相比具有意想不到的有益作用。本发明提供的多肽化合物化学性质稳定,不易被体内的dpp-iv和nep降解,不易被肾小球滤过,化合物的稳定性显著提高,具有支持每周一次给药的药代动力学特征。本发明提供的多肽化合物与已上市的glp-1受体激动剂和已报道的glp-1/胃泌素受体双重激动剂相比具有更低的免疫原性特性,更优异的降血糖活性、减重、调脂和降低肝脏脂质含量作用,对t2dm、肥胖、nafld、nash等代谢性疾病的治疗作用优于现有上市药物和已报道的glp-1/胃泌素受体双重激动剂。因此,本发明提供的多肽化合物,适合作为治疗代谢性疾病,如糖尿病、肥胖、nafld、nash等药物的活性成分。
附图说明
[0029]
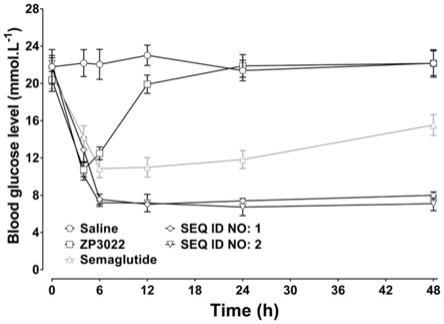
图1显示的是各受试物单次给药在db/db小鼠非禁食状态下的长效降血糖作用;
[0030]
图2显示的是各受试物在体外的免疫原性。
具体实施方式
[0031]
在本说明书全文中采用以下缩写:
[0032]
英文缩写
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
中文
[0033]
aib
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
α-氨基异丁酸
[0034]
ala
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
丙氨酸
[0035]
asp
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
天冬氨酸
[0036]
asn
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
谷氨酰胺
[0037]
glu
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
谷氨酸
[0038]
gly
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
甘氨酸
[0039]
his
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
组氨酸
[0040]
ile
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
异亮氨酸
[0041]
leu
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
亮氨酸
[0042]
lys
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
赖氨酸
[0043]
phe
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
苯丙氨酸
[0044]
thr
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
苏氨酸
[0045]
tyr
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
酪氨酸
[0046]
trp
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
色氨酸
[0047]
val
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
缬氨酸
[0048]
styr
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
磺基化酪氨酸
[0049]
nle
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
芴甲氧羰酰基正亮氨酸
[0050]
aeea
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
8-氨基-3,6二氧杂辛酸
[0051]
dcm
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二氯甲烷
[0052]
dmf
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二甲基甲酰胺
[0053]
fmoc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
9-芴基甲氧基羰基
[0054]
boc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
叔丁氧羰基
[0055]
dmso
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二甲基亚砜
[0056]
dic
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
n,n
’‑
二异丙基碳二亚胺
[0057]
hobt
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1-羟基-苯并三氮唑
[0058]
alloc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
烯丙氧羰基
[0059]
dde
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1-(4,4-二甲基-2,6-二氧代亚环己基)-乙基
[0060]
mtt
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
4-甲基三苯甲基
[0061]
ivdde
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1-(4,4-二甲基-2,6-二氧代亚环己基)3-甲基-丁基
[0062]
tfa
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
三氟乙酸
[0063]
edt
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二巯基乙烷
[0064]
hplc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
高效液相色谱
[0065]
lc-ms
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
液质联用质谱
[0066]
dmem
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
杜贝可氏修饰伊格氏培养基
[0067]
fbs
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
胎牛血清
[0068]
pbs
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
磷酸盐缓冲盐水
[0069]
hepes
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2-[4-(2-羟乙基)哌嗪-1-基]乙磺酸
[0070]
bsa
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
牛血清白蛋白
[0071]
ibmx
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
3-异丁基-1-甲基黄嘌呤
[0072]
hbss
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
hanks’平衡盐溶液
[0073]
aimv
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
无血清细胞培养基
[0074]
实施例1
[0075]
多肽化合物的合成(seq id no:1)
[0076][0077]
(1)树脂的溶胀
[0078]
称取担载量为0.382mmol/g的rink amide mbha树脂0.262g(0.1mmol当量),放入25ml的反应器中,用7ml的dcm和甲醇交替清洗树脂1次,7ml的dcm清洗树脂2次,然后用7ml的dcm溶胀树脂1h,最后用7ml dmf清洗树脂3次。
[0079]
(2)树脂fmoc保护基的脱除
[0080]
将溶胀后的树脂转入psi200多肽合成仪,加入7ml 20%哌啶/dmf(v/v)室温反应5min,滤去脱保护溶液,7ml dmf清洗树脂一次,再加入7ml 20%哌啶/dmf(v/v)脱保护溶剂与树脂反应15min,最后7ml dmf清洗树脂4次,每次1.5min,得到脱除fmoc保护基的rink树脂。
[0081]
(3)fmoc-phe-rink amide-mbha resin的合成
[0082]
称fmoc-phe-oh(0.4mmol),用3ml 10%dmf/dmso(v/v)溶解,加入2ml dic/hobt(0.4mmol/0.44mmol)缩合剂,预活化30min后,将活化好的氨基酸加入反应器中,室温震荡反应2h,滤去反应液后用7ml dmf清洗树脂4次,使用kaiser试剂检测反应耦合是否完全,如不完全则2次耦合。
[0083]
(4)肽链的延长
[0084]
按照肽链的序列,重复上述脱保护和耦合的步骤依次连接上相应的氨基酸,直至肽链合成完毕。其中styr侧链磺酸基使用新戊基保护,采用fmoc-tyr(so
3-neopentyl)-oh。侧链修饰的lys可以采用fmoc-lys(alloc)-oh、fmoc-lys(dde)-oh、fmoc-lys(mtt)-oh或fmoc-lys(ivdde)-oh等。本实例中采用fmoc-lys(dde)-oh保护策略,同时n末端的his使用的是boc-his(boc)-oh。
[0085]
(5)lys侧链的修饰
[0086]
肽链合成完毕后,加入7ml 2%水合肼/dmf(v/v)选择性脱除lys的dde保护基,dde保护基脱除后加入0.4mmol的fmoc-aeea-oh,0.4mmol的dic及0.44mmol的hobt,震荡缩合反应2h。脱除fmoc保护基后,再次加入0.4mmol的fmoc-aeea-oh,0.4mmol的dic及0.44mmol的hobt,震荡缩合反应2h。脱除fmoc保护基后,加入0.4mmol的fmoc-glu-otbu,0.4mmol的dic及0.44mmol的hobt,震荡缩合反应2h。脱除fmoc保护基后,加入0.4mmol的十八烷二酸单叔丁酯或二十烷二酸单叔丁酯,0.4mmol的dic及0.44mmol的hobt缩合反应2h,反应完全后用7ml dmf清洗树脂4次。
[0087]
(6)多肽的裂解
[0088]
将上述得到的连有多肽的树脂转移至圆底瓶中,使用切割剂reagent r(tfa/苯甲硫醚/苯酚/edt,90:5:3:2,v/v)5ml切割树脂,在油浴中恒温30℃反应2h,切割液倾入40ml冰乙醚中,冷冻离心后粗品用15ml冰乙醚洗涤3次,最后用氮气吹干,得到粗肽。
[0089]
(7)styr侧链脱除新戊基及多肽的纯化
[0090]
粗肽在乙腈醋酸铵水溶液中55℃搅拌反应12小时,脱除新戊基。脱除新戊基后,反应溶液直接用0.25μm微孔滤膜过滤后进岛津制备型反相hplc系统纯化。色谱条件为c18反相制备柱(250mm
×
20mm,12μm);流动相a:0.1%tfa/水(v/v),流动相b:甲醇(v/v);流速为8ml/min;检测波长为214nm。采用线性梯度(20%b~80%b/30min)洗脱,收集目标峰,除去甲醇后冻干得纯品0.28g,纯度大于99%,通过lc-ms确认目标多肽的分子量。
[0091]
实施例2
[0092]
多肽化合物的合成(seq id no:2)
[0093][0094]
合成方法同实施例1,收集目标峰冻干得纯品0.25g。
[0095]
实施例3
[0096]
多肽化合物对glp-1受体、cck-1受体和cck-2受体的激动活性测定
[0097]
通过功能测定法来确定多肽化合物对受体的激动作用,glp-1受体激动活性所述测定法测量稳定表达人glp-1受体的hek-293细胞系的camp响应。将稳定表达glp-1受体的细胞分入t175培养瓶并在培养基(dmem/10%fbs)中过夜生长至接近汇合状态,然后除去培养基,并用无钙和镁的pbs洗涤细胞,然后用accutase酶进行蛋白酶处理。洗涤脱离的细胞并将其重悬于测定缓冲液(20mm hepes,0.1%bsa,2mm ibmx,1
×
hbss)中,并确定细胞密度,并将25μl的等分试样分装至96孔板的孔中。为了测量,将25μl的测试多肽化合物在测定缓冲液中的溶液添加到孔中,然后室温温育30分钟。用cisbio的试剂盒,基于均相时间分辨荧光(htrf)来确定细胞的camp含量。添加稀释于裂解缓冲液(试剂盒组分)中的htrf试剂
后,将平板温育1小时,然后测量665/620nm处的荧光比。通过检测引起最大响应的50%激活的浓度(ec
50
)来对激动剂的体外效力进行量化。
[0098]
稳定表达cck-1受体或cck-2受体的1321-n1细胞用dmem-31966(含有10%fbs,1%丙酮酸钠,1%青霉素,1%链霉素)培养。试验前一天,将细胞转移至384孔板中,化合物溶解在ip-one缓冲溶液中(含有10mmol/l hepes,1mmol/l cacl2,4.2mmol/l kcl,146mmol/l nacl,5.5mmol/l葡萄糖,50mmol/l licl)并稀释,加入384孔板中。在37℃孵育1小时后,使用ip-one htrf assay kit测定细胞内的1-磷酸肌醇浓度,通过检测引起最大响应的50%激活的浓度(ec
50
)来对激动剂的体外效力进行量化。
[0099]
将本专利申请实施例中的检测数据(nm)显示于下表1中,虽然用一定数量的有效数字来陈述检测数据,但不应该认为表示数据已确定精确为有效数字的数。
[0100]
表1:多肽化合物对glp-1受体、cck-1受体和cck-2受体(胃泌素受体)的ec
50
值(以nm表示)
[0101][0102][0103]
如表1所示,所有多肽化合物对glp-1受体的激动活性都高于天然glp-1,并且所有多肽化合物对cck-2受体的激动活性也高于cck-2受体的天然配体胃泌素-6,同时所有多肽化合物都表现出了cck-2受体激动的高选择性,选择性明显优于胃泌素-6。
[0104]
实施例4
[0105]
多肽化合物对dpp-iv和nep酶的稳定性
[0106]
受试样品于37℃与纯化的人dpp-iv或nep酶共孵0,2,4,8小时,使用hplc法测定各时间点溶液中的残留样品峰面积,计算样品半衰期,结果如表2所示。
[0107]
表2:多肽化合物在dpp-iv酶或nep酶体系中的半衰期(以h表示)
[0108]
样品半衰期(dpp-iv中)半衰期(nep中)glp-11.32.1胃泌素-60.91.1seq id no:1》8》8seq id no:2》8》8
[0109]
如表2结果显示,本发明的多肽化合物在含dpp-iv酶溶液和nep酶溶液体系中的半衰期均超过8个小时,优于天然glp-1和胃泌素-6,说明可以有效耐受dpp-iv和nep酶的降解。
[0110]
实施例5
[0111]
多肽化合物在大鼠体内的药代动力学性质
[0112]
大鼠给予50nmol/kg的皮下(s.c.)注射给药,在给药后0.25,0.5,1,2,4,8,16,24,
36和48小时收集血样。使用乙腈沉淀蛋白质后,用lc-ms分析血浆样品。用winonlin 5.2.1(非房室模型)计算药代参数和半衰期(表3)。
[0113]
表3:多肽化合物在大鼠体内的药代动力学概貌
[0114]
样品t
1/2
(h)c
max
(ng/ml)zp30221.0425semaglutide8.2481seq id no:114.3547seq id no:216.7561
[0115]
如表3结果显示,本发明的多肽化合物的体内半衰期显著延长,显著优于一周一次给药的semaglutide,并且也显著优于zp3022,具有支持每周一次给药的药代动力学特征。
[0116]
实施例6
[0117]
多肽化合物对糖尿病模型小鼠(db/db小鼠)血糖的影响
[0118]
雄性db/db小鼠,随机分组,每组6只。空白组皮下注射给予生理盐水(10mg/kg),给药组分为6组,小鼠实验期间自由进食和饮水,小鼠非空腹状态下分别皮下单次注射25nmol/kg的zp3022,semaglutide,seq id no:1,seq id no:2。在给药前0h,以及给药后4,6,24和48h用血糖仪测量各组小鼠血糖水平。
[0119]
如图1结果所示,在db/db小鼠体内的降血糖实验结果表明,本发明的多肽化合物显示出了显著优于阳性对照药semaglutide和zp3022的长效降血糖活性。
[0120]
实施例7
[0121]
多肽化合物对饮食诱导肥胖(dio)小鼠血糖、体重和血脂的影响
[0122]
雄性c57bl/6j小鼠,体重22g左右,模型组共42只,用research diets公司的d12492高脂饲料饲养18周造dio小鼠模型。在给药开始前,各组dio小鼠按照体重随机分组,共分为5组,每组6只,分别为生理盐水组(对照高脂饮食组)、阳性对照组(zp3022和semaglutide)和受试样品组(seq id no:1,seq id no:2)。对照高脂饮食组每天三次皮下注射生理盐水(10mg/kg),zp3022(25nmol/kg)每天三次皮下注射,semaglutide,seq id no:1,seq id no:2(剂量都为25nmol/kg)每两天注射一次,给药周期21天。每天记录小鼠体重变化,实验开始前和结束时使用核磁共振(nmr)来测量体脂量,使用血糖仪测量空腹血糖。在实验结束后,各组小鼠取血,测量血清中甘油三酯和胆固醇含量。最后,各组小鼠取肝脏,匀浆后提取肝脏脂质,测量肝脏的甘油三酯和胆固醇含量。
[0123]
表4:dio小鼠在3周给药周期内的体重、体脂和空腹血糖变化(%)
[0124]
样品整体体重变化(%)体脂变化(%)空腹血糖变化(%)对照高脂饮食 1.2%(
±
0.6%) 2.3%(
±
1.0%) 2.8%(
±
1.1%)zp3022-16.2%(
±
1.1%)
***-17.4%(
±
0.8%)
***-19.5%(
±
1.4%)
***
semaglutide-15.7%(
±
0.8%)
***-18.6%(
±
1.3%)
***-17.2%(
±
1.6%)
***
seq id no:1-32.8%(
±
4.1%)
***,###-39.8%(
±
3.1%)
***,###-34.6%(
±
2.6%)
***,###
seq id no:2-37.9%(
±
2.7%)
***,###-43.6%(
±
4.3%)
***,###-38.2%(
±
4.1%)
***,###
[0125]
***
:与对照高脂饮食组相比p《0.001;
###
:与zp3022和semaglutide组比p《0.001
[0126]
如表4结果显示,本发明的多肽化合物在dio小鼠体内连续给药3周,可以显著降低小鼠的体重和体脂含量,降低空腹血糖值,并且本发明的多肽化合物的作用显著强于阳性
对照药zp3022和semaglutide。
[0127]
表5:dio小鼠在3周给药周期后的血脂数据(以mmol/l表示)
[0128]
样品甘油三酯胆固醇对照高脂饮食1.26
±
0.072.3
±
0.2zp30221.02
±
0.08
***
1.7
±
0.1
***
semaglutide1.03
±
0.06
***
1.8
±
0.2
***
seq id no:10.71
±
0.08
***,##
1.0
±
0.2
***,##
seq id no:20.66
±
0.07
***,##
0.8
±
0.1
***,##
[0129]
***
:与对照高脂饮食组相比p《0.001;
##
:与zp3022和semaglutide组比p《0.01
[0130]
如表5结果显示,本发明的多肽化合物在dio小鼠体内连续给药3周,可以显著降低小鼠的血清甘油三酯和胆固醇含量,并且本发明的多肽化合物的作用显著强于阳性对照药zp3022和semaglutide。
[0131]
表6:dio小鼠在3周给药周期后的肝脏脂质数据(以mg/g肝脏表示)
[0132]
样品甘油三酯胆固醇对照高脂饮食94.6
±
6.89.4
±
0.8zp302276.8
±
4.1
***
7.4
±
0.5
***
semaglutide71.7
±
5.1
***
7.0
±
0.3
***
seq id no:146.8
±
2.8
***,##
5.6
±
0.4
***,##
seq id no:241.6
±
3.7
***,##
5.2
±
0.2
***,##
[0133]
***
:与对照高脂饮食组相比p《0.001;
##
:与zp3022和semaglutide组比p《0.01
[0134]
如表6结果显示,本发明的多肽化合物在dio小鼠体内连续给药3周,可以显著降低小鼠的肝脏甘油三酯和胆固醇含量,并且本发明的多肽化合物的作用显著强于阳性对照药zp3022和semaglutide。
[0135]
实施例8
[0136]
多肽化合物对db/db小鼠糖化血红蛋白(hba1c)、空腹血糖和胰岛面积的影响
[0137]
雄性db/db小鼠,随机分组,每组6只。适应性饲养一周后,尾部取血测量治疗开始前初始hba1c(%)数值和空腹血糖数值。空白组每天三次皮下注射给予生理盐水(10mg/kg),给药组分为4组,分别皮下注射25nmol/kg的zp3022(每天三次),semaglutide(每两天一次),seq id no:1(每两天一次),seq id no:2(每两天一次)。治疗周期为5周,治疗结束后小鼠禁食过夜后测量空腹血糖数值,同时取血测量hba1c(%)数值。最后小鼠处死,取胰腺做切片,he染色后在10倍镜下测量各组小鼠的胰岛面积。
[0138]
表7:db/db小鼠在5周给药周期内的hba1c和空腹血糖变化(%)
[0139]
样品(剂量)hba1c变化(%)空腹血糖变化(%)生理盐水 2.87%(
±
1.18%) 9.27%(
±
0.90%)zp3022-4.16%(
±
0.83%)
***-6.07%(
±
0.97%)
***
semaglutide-4.27%(
±
0.80%)
***-5.39%(
±
0.70%)
***
seq id no:1-9.48%(
±
0.46%)
***,##-16.27%(
±
2.11%)
***,##
seq id no:2-10.38%(
±
0.31%)
***,##-18.49%(
±
1.87%)
***,##
[0140]
***
:与对照高脂饮食组相比p《0.001;
##
:与zp3022和semaglutide组比p《0.01
[0141]
如表7结果显示,本发明的多肽化合物在db/db小鼠体内连续给药5周,可以降低hba1c和空腹血糖,显著优于阳性对照药zp3022和semaglutide,说明具有很好的血糖控制作用。
[0142]
表8:db/db小鼠在5周给药后的胰岛面积(以μm2表示)
[0143]
样品(剂量)胰岛面积生理盐水21296
±
1847zp302229698
±
1530
***
semaglutide28156
±
1357
***
seq id no:142598
±
1876
***,###
seq id no:245782
±
1916
***,###
[0144]
***
:与生理盐水组相比p《0.001;
###
:与liraglutide和semaglutide组比p《0.001
[0145]
如表8结果显示,本发明的多肽化合物在db/db小鼠体内连续给药5周,可以显著增加db/db小鼠的胰岛面积,说明具有很高的促进胰腺组织的细胞增殖和胰岛再生的作用,并且本发明的多肽化合物的作用显著强于阳性对照药zp3022和semaglutide。
[0146]
实施例9
[0147]
多肽化合物的免疫原性
[0148]
采用来自50例中国人捐赠者的外周血单个核细胞(pbmc)进行了诱导t细胞增殖的免疫原性实验。pbmc在aimv培养基中培养,并添加到24孔板(2ml)中以达到最终浓度~3
×
106cells/ml,然后通过在aimv培养基中添加zp3022,semaglutide,6a(选自j.med.chem.2020,63,12595-12613),seq id no:1,seq id no:2来刺激pbmc。24孔板在37℃的二氧化碳培养箱(5%)中培养8天。第5天、第6天、第7天和第8天,将培养板各孔的细胞转移到96孔板上。用[3h]-胸腺嘧啶核苷对培养物进行处理,再培养18小时,并测定每个孔的每分钟计数(cpm)。刺激指数(si)是通过将每个供体的试验孔的增殖反应(cpm)除以培养基处理(cpm)的增殖反应来计算的,大于2.0的si被视为阳性。通过将整个时间过程(5-8天)内有阳性反应的捐赠者数量占接受测试的捐赠者总数的百分比来计算捐赠者的响应百分比。
[0149]
如图2结果所示,本发明的多肽化合物的捐赠者反应比例显著低于zp3022,6a和semaglutide,说明本发明的多肽化合物具有更低的免疫原性。
[0150]
最后所应说明的是,以上具体实施方式仅用以说明本发明的技术方案而非限制,尽管参照实例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
