一种枯草芽孢杆菌全长cdna文库构建方法及其定向筛选应用
技术领域
1.本发明涉及一种枯草芽孢杆菌全长cdna文库构建方法及其定向筛选应用,属于基因工程技术领域。
背景技术:
2.cdna文库是特定物种或组织某一时期所有转录物信息的集合,以这些转录物为模板反转录合成的cdna经过与特定的载体连接可以获得含有这些转录物信息的克隆集合。这些集合可以凭借二代测序技术用于转录组测序,rna结构探查,mrna剪接位点寻址等,或是转化受体菌用于功能基因的鉴定或蛋白互作分析。
3.枯草芽孢杆菌(bacillus subtilis)是芽孢杆菌属(bacillus cohn)的一种革兰氏阳性菌株。b.subtilis由于不产毒素和致热致敏蛋白,因此被列为美国fda《gras(公认为安全物质)》清单认证菌株,其易于分离培养,可高密度发酵和蛋白分泌能力强的特性,使得枯草芽孢杆菌作为重要的工业菌株,用于生产多种外源重组蛋白和代谢产物,其优秀性状具备较高的鉴定价值。但是由于枯草芽孢杆菌等原核生物mrna结构缺乏类似真核生物的polya尾,导致通过oligo dt引物起始的反转录反应无法进行,而原核生物cdna文库构建中常用的随机引物方法则无法有效获得全长cdna。由于对于枯草芽孢杆菌cdna文库构建方法的描述较少,现有的枯草芽孢杆菌cdna文库构建研究仅在karnik p,gopalakrishna y,sarkar n.construction of a cdna library from polyadenylated rna of bacillus subtilis and the determination of some 3
′‑
terminal sequences[j].gene,1986,49(1):161-165.和gopalakrishna y,sarkar n.the synthesis of dna complementary to polyadenylate-containing rna from bacillus subtilis.[j].journal of biological chemistry,1982,257(6):2747-50.中被描述。这些研究均以枯草芽孢杆菌总rna中的polya
mrna为模板通过oligo dt引物介导反转录反应,然而,poly a
mrna仅占总mrna的约40%,这大大限制了转录本的丰富性,导致无法生成具有代表性的cdna文库。因此,通过构建原核生物cdna文库鉴定其功能基因存在一定限制,其高质量cdna文库的构建未见报道。
技术实现要素:
[0004]
本发明提供了一种b.subtilis全长cdna文库构建方法及定向筛选应用,以解决上述技术问题。
[0005]
本发明的第一个目的是提供一种构建b.subtilis全长cdna文库的方法,所述方法包括以下步骤:
[0006]
s1:提取枯草芽孢杆菌的总rna,并去除总rna中的rrna;
[0007]
s2:对s1中去除rrna的总rna进行体外腺苷酸化并进行纯化获得poly a
rna;
[0008]
s3:利用具有末端转移活性的逆转录酶将s2中的poly a
rna反转录成单链cdna,并用与逆转录酶末端转移活性生成碱基互补配对的寡合苷酸标记单链cdna的3’末端;
[0009]
s4:将s3中的单链cdna在pcr引物和锚定引物的引导下合成并扩增cdna第二条链,
纯化富集片段大小≥500bp的双链cdna;
[0010]
s5:将s4中的双链cdna连接到质粒上并转化至大肠杆菌,获得b.subtilis全长cdna文库。
[0011]
在本发明的一种实施方式中,s1中的提取总rna的方法可以是柱式提取法。
[0012]
在本发明的一种实施方式中,s1中取出rrna的方法为利用细菌rrna去除试剂盒。
[0013]
在本发明的一种实施方式中,s2中采用poly(a)聚合酶进行体外腺苷酸化反应,采用mrna纯化磁珠进行纯化。
[0014]
在本发明的一种实施方式中,s3中的反转录的引物的序列如seq id no.1所示。
[0015]
在本发明的一种实施方式中,s4中的pcr引物的序列如seq id no.2所示,锚定引物的序列如seq id no.3所示。
[0016]
在本发明的一种实施方式中,s4中利用凝胶层析柱纯化富集双链cdna。
[0017]
在本发明的一种实施方式中,s5中所述质粒包括pad123。
[0018]
本发明的第二个目的是提供一种b.subtilis筛选系统,所述筛选系统以枯草芽孢杆菌为宿主,包含外源基因的筛选质粒和根据上述方法构建的b.subtilis全长cdna文库抽提得到的混合质粒。
[0019]
在本发明的一种实施方式中,所述筛选质粒以pub110为载体。
[0020]
在本发明的一种实施方式中,所述外源基因包括但不限于gfp基因或酶。
[0021]
在本发明的一种实施方式中,所述枯草芽孢杆菌为枯草芽孢杆菌sck6。
[0022]
本发明的第三个目的是提供一种构建b.subtilis筛选系统的方法,将上述方法中制备得到的b.subtilis全长cdna文库进行质粒抽提,并转化至含有携带外源基因的筛选质粒的枯草芽孢杆菌。
[0023]
在本发明的一种实施方式中,所述筛选质粒以pub110为载体。
[0024]
在本发明的一种实施方式中,所述外源基因包括但不限于gfp基因或其他模型蛋白基因。
[0025]
在本发明的一种实施方式中,所述枯草芽孢杆菌为枯草芽孢杆菌sck6。
[0026]
有益效果:
[0027]
1.本发明的方法可以构建得到高质量b.subtilis全长cdna文库,库容量达到7.5
×
103cfu/ml以上;以制备的单链cdna为模板,检测表达丰度不同的六个b.subtilis管家基因gapa、rple、ssba、rpob、mreb和rpso的扩增效率达到100%,为其他原核生物cdna文库构建提供参考。
[0028]
2.本发明构建的筛选系统可以直接用于枯草芽孢杆菌功能基因的定向筛选以及关键基因的鉴定。
附图说明
[0029]
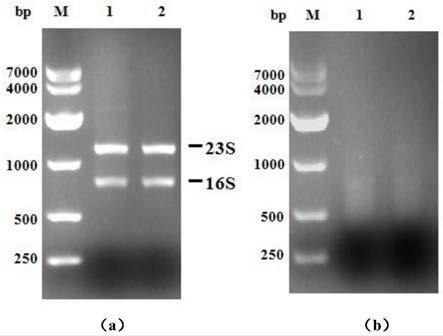
图1为总rna及去除rrna后的总rna电泳检测图;(a)总rna电泳检测,(b)去除rrna后的总rna电泳检测图。
[0030]
图2为内参基因的信息与核酸电泳检测图;(a)内参基因的信息,(b)核酸电泳检测图。
[0031]
图3为双链cdna合成循环数优化及分级电泳检测图;(a)合成循环数优化,(b)大片
段双链cdna电泳检测图。
[0032]
图4为pad123-p
glv
载体表达cdna质粒示意图。
[0033]
图5为pub110载体表达gfp质粒示意图。
[0034]
图6为pad123与pub110质粒信息示意图。
[0035]
图7为插入片段pcr验证核酸电泳检测图。
具体实施方式
[0036]
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。
[0037]
除非特别说明,以下实施例所用试剂和材料均为市售商品或者可以通过已知方法制备。
[0038]
下述实施例中涉及到的质粒:
[0039]
1、穿梭质粒pad123-p
glv
:pad123质粒骨架取自dunn a k,handelsman j.a vector for promoter trapping in bacillus cereus.[j].gene,1999,226(2):297-305.中所报道的穿梭质粒。通过同源重组将gfpmut3基因替换为核苷酸序列如seq id no.6所示的麦芽糖诱导型启动子p
glv
,使用引物如下:
[0040]
fp
glv
:
[0041]
5'-tttaagaaggagatatacatggcatgtatccgaatcgtacaaaag-3'
[0042]
rp
glv
:
[0043]
5'-cttgcatgcctgcaggagatgaattcacctccttgataaatttatttatttaagatc-3'
[0044]
fpad:
[0045]
5'-atctcctgcaggcatgcaag-3'
[0046]
rpad:
[0047]
5'-atgtatatctccttcttaaatctagaggatccc-3'
[0048]
2、质粒pub110-gfp:pub110质粒骨架取自keggins km,lovett ps,duvall ej.molecular cloning of genetically active fragments of bacillus dna in bacillus subtilis and properties of the vector plasmid pub110.proc natl acad sci u s a.1978;75(3):1423-1427.中报道的质粒。通过同源重组将核苷酸序列如seq id no.7所示的p
amyq
’-gfp连接到pub110载体,使用引物如下:
[0049]
fgfp:
[0050]
5'-accgaagcagaaacagaacgccgccgatccaggagaacaaaaacgattttg-3'
[0051]
rgfp:
[0052]
5'-agcttggaggtgtttttttattaccttatttgtatagttcatccatgccatgtg-3'
[0053]
fpub:
[0054]
5'-tggcatggatgaactatacaaataaggtaataaaaaaacacctccaagctgag-3'
[0055]
rpub:
[0056]
5'-atgtatatctccttcttaaatctagaggatccc-3'
[0057]
下述实施例中涉及的培养基:
[0058]
培养基均使用ddh2o配制,配制完成后121℃灭菌15~20min。
[0059]
lb液体培养基:酵母粉5.0g/l、胰蛋白胨10.0g/l、nacl 10.0g/l。
[0060]
lb固体培养基:酵母粉5.0g/l、胰蛋白胨10.0g/l、nacl 10.0g/l、琼脂粉18g/l。
[0061]
yn液体培养基:酵母粉7.0g/l、营养肉汤18g/l。
[0062]
实施例1构建枯草芽孢杆菌cdna文库
[0063]
(1)总rna的提取及rrna去除。
[0064]
将枯草芽孢杆菌b.subtilis 168接种于50ml体系的lb培养基中培养6.5h至对数生长期,采用柱式rna提取试剂盒(takara)提取b.subtilis 168菌株总rna,2.5%凝胶电泳检测rna条带清晰,23s rrna条带的亮度大约是16s rrna的两倍(图1a),采用thermo nanodrop 2000核酸分析仪检测总rna浓度为141.23ng/μl,a260/a280为2.08,a260/a230为1.93,总体积200μl,rna总量为28.25μg。
[0065]
采用细菌rrna去除试剂盒(neb)去除总rna中的rrna,按照表1配置体系:
[0066]
表1
[0067]
加样组分加样量总rna11μlrrna depletion solution2μlprobe hybridization buffer2μl
[0068]
混匀后于95℃反应2min,以0.1℃/min缓慢降至22℃,继续反应5min,在反应后的体系中加入表2中的组分:
[0069]
表2
[0070]
加样组分加样量rnase h reaction buffer2μlrnase h2μlnuclease-free水1μl
[0071]
混匀后50℃反应30min,在反应后的体系中加入表3中的组分:
[0072]
表3
[0073]
加样组分加样量dnase i reaction buffer5μldnase i2.5μlnuclease-free水22.5μl
[0074]
混匀后37℃反应30min,加入试剂盒中90μl纯化磁珠至反应结束后的体系,混匀,静置,弃上清,并加入200μl 80%乙醇重悬,静置,弃上清,空气干燥5min,加入7μlnuclease-free水重悬,静置,分离上清并收集,2.5%凝胶核酸电泳检测去除rrna的总rna电泳图结果显示rrna特征带消失(图1b)。
[0075]
(2)rna体外腺苷酸化和分离纯化。
[0076]
采用escherichia coli poly(a)聚合酶(neb)进行体外腺苷酸化反应,取步骤(1)中的去除rrna的总rna 37.5μl,按表4配置反应体系:
[0077]
表4
[0078]
加样组分加样量
rna37.5μl10
×
poly(a)buffer5μlatp(10mm)5μlpoly(a)polymerase2.5μl
[0079]
混匀后37℃反应30min,反应结束后直接进行纯化,采用mrna纯化磁珠(vazyme)富集polya
rna,采用thermo nanodrop 2000核酸分析仪检测总rna浓度为36.56ng/μl,a260/a280为2.21,a260/a230为2.05,总体积10μl,polya
rna总量为365.6ng。
[0080]
(3)cdna第一条链合成
[0081]
采用cdna文库构建试剂盒(takara)可以在反转录的同时并为合成全长cdna标记3’末端。取步骤(2)中的poly a
rna,按照表5配置反应体系:
[0082]
表5
[0083]
加样组分加样量poly a
rna3μl3’in-fusion smarter cds primer(12μm)(seq id no.1)1μl去离子水0.5μl
[0084]
混匀后72℃反应3min,42℃反应2min后,加入按照表6配置的主混液,总体系10μl,42℃反应90min,68℃反应10min终止反应,制备得到单链cdna。
[0085]
表6
[0086][0087][0088]
选取表达丰度不同的六个b.subtilis管家基因gapa、rple、ssba、rpob、mreb和rpso为内参设计引物(图2a),基因的表达丰度跨度由5.3至100.0rpkm,取单链cdna0.5μl作为pcr模板,进行pcr扩增并利用1%凝胶电泳检验单链cdna质量(图2b),所有内参基因均有效扩增,扩增率达到100%,且片段大小符合预期,表明反转录得到的单链cdna质量较高,在转录物丰富度与完整度上显著优于已有的枯草芽孢杆菌cdna文库。
[0089]
(4)cdna第二条链合成及分级分离。
[0090]
采用advantage hd pcr聚合酶(takara)合成cdna第二条链,取步骤(3)中的单链cdna2μl作为pcr模板,按照表7配置4个平行反应体系:
[0091]
表7
[0092]
加样体系加样量单链cdna2μl
去离子水71μl5
×
advantage hd buffer20μl50
×
dntp mix(10mm)2μl5’pcr primer ii a(12μm)(seq id no.2)2μl3’in-fusion smarter pcr primer(12μm)(seq id no.3)2μladvantage hd polymerase mix1μl
[0093]
混匀后按照表8设置pcr反应条件:
[0094]
表8
[0095][0096][0097]
将配制的反应体系进行15次pcr循环,15次pcr循环结束后取出5μl反应液作为后续凝胶核酸电泳样本,从剩余反应体系吸取25μl继续进行pcr反应,分别取循环数为18、21、24、27的反应样品各5μl,每次取出5μl样品后剩余反应体系放回pcr仪继续循环。27次循环反应结束后,将收集的各个循环数的5μl样品进行1%凝胶核酸电泳,结果如图3a所示,当循环数为15时,弥散的cdna混合物能被显著观察到,随着循环数的上升,弥散带的位置不断上移,这是因为再扩增过程中形成了多聚体,因此确定最佳反应循环数为15循环(图3a),在每100μl pcr体系中加入11.5μl 1%二甲苯青ff染料,经chroma spin te-1000凝胶层(takara)析柱收集单滴馏分富集500bp以上的大片段双链cdna,取5μl单滴馏分进行1%凝胶核酸电泳(图3b)。
[0098]
将收集的双链cdna用冷乙醇沉淀,干燥后用去20μl离子水复溶,采用thermo nanodrop 2000核酸分析仪检测双链cdna浓度为50ng/μl,a260/a280为1.79。证明获得的双链cdna纯度较高。
[0099]
(5)同源重组与转化宿主细胞。
[0100]
以穿梭质粒pad123-p
glv
表达步骤(4)中的双链cdna。根据质粒pad123-p
glv
的质粒图谱设计用于制备线性化载体pad123-p
glv
的引物(图4),引物序列如下:
[0101]
p3:(seq id no.4)
[0102]
5'
–
tctcatcgtaccccgatctcctgcaggcatgcaag
–
3'
[0103]
p5:(seq id no.5)
[0104]
5'
–
ttgataccactgcttgaattcacctccttgataaatttatttatttaagatcc
–
3'
[0105]
按照表9配置2个同源重组连接体系:
[0106]
表9
[0107]
加样组分加样量双链cdna(50ng/μl)6μl线性化载体pad123-p
glv
(150ng/μl)2μl5
×
in-fusion hd enzyme premix2μl
[0108]
混匀后50℃反应15min,加入90μl te buffer,10μl quick clean树脂,混匀后离心收集上清,加入1.2μl糖原及280μl 100%乙醇,过夜沉淀,15000rpm离心20min去除上清,空气干燥后用10μl去离子水重悬得连接产物。将5μl连接产物于冰上加入50μl hst08感受态细胞,共转化4支感受态细胞,电压设置1500v,电击后立即加入1ml预冷的soc培养基,37℃,200rpm复苏1h,取5μl菌液稀释涂布在含有氨苄青霉素抗性(100μg/ml)的lb平板上,过夜培养,统计菌落数。将平板上的菌落全部收集,提取混合质粒,取10μl转化至含有pub110-gfp表达质粒的b.subtilis sck6(图5)感受态细胞,冰浴20min,37℃水浴20min,37℃,200rpm复苏3h,涂布在含有卡那霉素抗性(40μg/ml)及氯霉素抗性(20μg/ml)的lb平板上,过夜培养,统计菌落数,双质粒在sck6受体菌中可以各自稳定表达,质粒的筛选抗性及复制原点信息如图6所示。
[0109]
实施例2文库质量评价及应用
[0110]
将实施例1中制备的表达双链cdna的pad123-p
glv
转化至hst08感受态细胞得hst08文库菌液,于1ml的总转化菌液中取5μlhst08文库菌液加入50μl lb液体培养基中,涂布于含有氨苄青霉素抗性(100μg/ml)的lb平板上,过夜培养,统计4个转化体系总克隆数,转化平板的菌液浓度为cfu/ml=46
×
200
×
4=3.68
×
104cfu/ml,随机挑取10个单菌落进行pcr验证(图7),按照如下体系配置pcr反应体系:
[0111]
表10
[0112]
加样组分加样量单菌落1去离子水23μlfcx(20μm)(seq id no.8)1μlrcx(20μm)(seq id no.9)1μl2
×
phanta mixture25μl
[0113]
反应条件如下:
[0114]
表11
[0115][0116]
[0117]
如图7所示,插入穿梭载体pad123-p
glv
的片段平均大小大于1.5kb,证明插入片段质量较高。
[0118]
刮取平板上所有的菌落至20ml液体lb培养基中,提取混合质粒并分别吸取10μl混合质粒转化至50管含有pub110-gfp表达质粒的sck6感受态细胞得sck6文库菌液,将sck6文库菌液涂布于含有卡那霉素抗性(40μg/ml)及氯霉素抗性(20μg/ml)的lb平板上,过夜培养,统计平板菌落数,50个平板上的平均菌落数为150个,最终计算得到转化平板的菌液浓度为cfu/ml=150
×
50=7.5
×
103cfu/ml。
[0119]
刮取平板上所有单菌落至lb液体培养基,加入5g
·
l-1
麦芽糖诱导cdna表达,培养至对数期后,将菌液稀释至适当菌浓,以含有pub110-gfp和未携带双链cdna的空质粒pad123-p
glv
的sck6重组菌作为阴性对照,流式分选gfp荧光信号增强的菌株,将荧光值增强的细胞群进行96孔板复筛和摇瓶验证,将摇瓶水平验证有效果的菌株提取质粒并测序,最终获得枯草芽孢杆菌中能够增强外源蛋白表达的功能基因。
[0120]
综上所述,本发明成功构建了在b.subtilis sck6中构建了b.subtilis全长cdna文库及用于筛选功能基因的双质粒筛选系统,不仅为原核生物cdna文库的建立方法提供了新的思路,同时也为cdna文库的后续应用奠定了基础。
[0121]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。