碳量子点@有机壳层结构的荧光纳米粒子的合成方法
1.技术领域:本发明涉及一种在空气中荧光稳定性良好的碳量子点@有机壳层结构的碳量子点的制备方法。
2.技术背景:碳量子点主要由c元素组成,与生物分子的相容性好。可通过溶剂热合成方法合成出发光效率较高的碳量子点发光材料,掺杂n、s等富电子元素可提高其荧光量子产率,可通过碳量子点表面基团的修饰来调控碳量子点的发光颜色。碳量子点的合成本较低、是具有良好的光致发光特性的纳米粒子。碳量子点荧光材料在生物成像、分子荧光检测、蛋白质检测等方面具有广阔的应用前景。目前蓝光、绿光、黄光和红光碳量子点的合成技术已经逐渐成熟,水热法、溶剂热法、微波法等合成方法得到了广泛的应用。文献报导的绿光和红光碳量子点的量子产率可达90%和80%。通常碳量子点的粒径在3-5nm,表面能很高,其荧光主要是由于碳量子点表面的缺陷能级产生的。碳量子点的表面缺陷能级在空气中易受氧气、水分子等的影响而发生荧光猝灭现象,限制了碳量子点在空气环境下的应用范围。申请人以异丙醇为碳源,以间苯二胺为氮源,以原位合成的sno2纳米粒子作为催化剂在4小时内高效地合成了量子产率为55.8%的绿光碳量子点(gcqds)。gcqds表面含有较多的-nh2基团,能够分散于水、乙醇、四氢呋喃等多种溶剂中,在溶液中具有良好的荧光稳定性(mater.todaycommun.2021,26:101762)。
3.蛋白质的磷酸化是生物代谢过程中的重要生命过程,是调节和控制蛋白质活力和功能的重要机制,与dna损伤修复、转录调节、信号传导等生物过程密切相关。异常的蛋白质磷酸化过程则会引发一系列疾病。因此分析和检测磷酸化蛋白在生命科学研究中是非常重要的。磷酸化蛋白质的检测主要用磷酸化蛋白的抗体免疫印迹法进行,然而磷酸化蛋白抗体的价格昂贵且不易保存,亟需一种生物相容性良好且特异性识别磷酸化蛋白质的荧光材料来代替磷酸化蛋白抗体的作用(j.chem.technol.biotechnol.2016,91:892
–
900)。因此合成在空气中荧光稳定且能够特异性识别磷酸化蛋白质的碳量子点具有重要的实用价值。
4.
技术实现要素:
本发明以提高碳量子点在空气中的荧光性能的稳定性为出发点,通过有机化学反应在绿光碳量子点粒子表面化学键合含有c=c双键的有机基团,通过自由基聚合反应在gcqds粒子表面聚合形成有机聚合物壳层,来保护绿光碳量子点的表面缺陷,以达到稳定碳量子点在空气中的荧光性能的目的。并且这种核@壳结构的碳量子点容易通过共聚反应键合上多羧酸功能基团,多羧酸功能基团通过螯合反应来抓取可以特异性识别磷酸化蛋白质分子的ti
4
、fe
3
等金属离子。
附图说明:
5.图1为本发明实施例一所得gcqd@ab-1的高分辨透射电子显微镜图(hrtem),插图为局部放大2.7倍的hrtem图。
6.图2为实施例一所得gcqds、gcqd@ab-1和实施例二所得gcqd@pcs在空气中放置不同时间的荧光稳定性图(在365nm紫外灯下拍摄)。
7.图3为实施例一所得gcqd@ab-1和gcqd-ab的核磁共振氢谱图。
8.图4为实施例一所得gcqd@ab-1的荧光激发和发射光谱图。
9.图5为本发明实施例二所得gcqd@pcs的hrtem图,插图为局部放大3倍的hrtem图。
10.图6为实施例二所得gcqd@pcs的荧光激发和发射光谱图。
11.图7为实施例三所得gcqd@ab-2的红外吸收光谱图。
12.图8为实施例三所得gcqd@ab-2的荧光激发和发射光谱图
13.为了提高碳量子点在空气中荧光性能的稳定性,设计在碳量子点粒子表面包裹一层有机聚合物,合成核@壳结构的碳量子点。利用烯丙基溴或对氯甲基苯乙烯等与gcqds表面的-nh2的反应,在碳量子点粒子表面引入c=c双键,然后通过c=c双键的自由基聚合反应,在碳量子点表面形成一层有机聚合物壳层,合成核@壳结构的碳量子点,设计合成路径如下。
14.合成路线1:
[0015][0016]
为了让gcqd@有机壳层结构的粒子抓取特异性识别磷酸化蛋白质的ti
4
等金属离子,设计合成可通过共聚反应在gcqd@有机壳层结构粒子的表面键合的聚合单体,n,n
’‑
二(乙酸乙酯基)烯丙基胺(dad)。dad合成路线2如下图所示。
[0017][0018]
然后再将dad通过自由基聚合反应,键合到gcqd@ab-1粒子的表面。合成路线3如下所示。
[0019][0020]
核@壳结构中的碳量子点不仅限于本发明中的gcqds,也可以是其它表面富含-nh2的碳量子点荧光纳米粒子。除了烯丙基溴和对氯甲基苯乙烯之外,也可以选择其它同时含有双键和卤素原子的物质作为双键的来源;聚合单体不仅限于苯乙烯,也可以选择烯丙基溴、甲基丙烯酸甲酯等作为聚合单体。
具体实施方式:
[0021]
以下结合具体的实施例子对上述方案做进一步说明,但不仅限于此。
[0022]
实施例一:
[0023]
参见合成路线1,核壳结构gcqd@ab-1碳量子点的合成包括以下步骤:
[0024]
a.绿光碳量子点gcqds的合成参考文献(mater.today commun.2021,26:101762)的方法合成。纯化后的gcqds的异丙醇溶液置于4℃冰箱中冷却保存。
[0025]
b.将gcqds溶液分散于10~50ml异丙醇与1~3ml去离子水的混合溶液中,并将其转移至三颈烧瓶。在搅拌状态下加入6~12mmol k2co3和5mg正四丁基溴化铵,通氮气30~60分钟。用注射器少量多次注入4~12mmol烯丙基溴,在80℃下回流反应,用薄层色谱监测反应进程,直至gcqds表面的-nh2反应完全。用旋蒸仪去除上述溶液中的溶剂,加入10~30ml异丙醇并超声溶解糖浆状产物(gcqd-ab),过滤除去不溶的k2co3等固体颗粒,得到10~30ml gcqd-ab的异丙醇溶液。1h nmr(400mhz,cdcl3):δ5.85(m,3j=4hz,1h),5.18(d,3j=12hz,1h),5.14(d,3j=8hz,1h),3.88(d,3j=4hz 2h)。1h nmr数据证实了gcqd-ab的合成。
[0026]
c.5mlgcqd-ab溶液用旋转蒸发仪去除溶剂,并将产物分散于12~30ml四氢呋喃中,加入2~5ml去离子水,超声10分钟。将溶液转移至三颈烧瓶,在搅拌状态下加入5~10mg聚乙烯吡咯烷酮,5mg偶氮二异丁腈,5mg正四丁基溴化铵。通氮气30分钟后,在64℃下反应24h。加入5mg 2,6-二叔丁基对甲酚终止聚合反应。最终得到核壳结构的碳量子点溶液gcqd@ab-1。
[0027]
图1是实施例一制备的gcqd@ab-1的高分辨透射电子显微镜图,纳米粒子均匀分散,且粒径约为3~5nm,每个gcqd@ab-1粒子表面形成了约1nm厚的有机聚合物壳层,表现出了gcqd@有机聚合物的核@壳结构。图1中的插图为局部放大2.7倍的hrtem电镜图,黑色近球形的是gcqd@ab-1的核,外层白色的圆环是有机聚合物壳层。
[0028]
gcqd@ab-1的荧光性能在空气中的稳定性实验。将gcqds溶液和本实施例一制备的gcqd@ab-1溶液用毛细管点到薄层色谱板上,置于空气中自然放置。图2是在365nm紫外灯照射下拍摄的置于空气中不同时间下的薄层色谱的图像。在空气中,gcqds的荧光信号逐渐减弱,5小时后其荧光基本消失。gcqd@ab-1在8天内(197小时)的荧光强度基本不衰减,8天之后其荧光信号才开始出现轻微衰减,12天后其荧光强度仍然保持较高的水平,说明核壳结构的gcqd@ab-1在空气中的荧光性能的稳定性远远高于gcqds。
[0029]
图3为本实施例一制备的gcqd-ab和gcqd@ab-1的核磁共振氢谱图。gcqd-ab的1hnmr(400mhz,cdcl3):δ5.85(m,3j=4hz,1h),5.16(d,d,3j=9.3hz,2h),3.88(d,3j=4hz,2h)。gcqd@ab-1:1h nmr(400mhz,cdcl3)δ3.58(s,2h),1.93(t,3j=4.00hz,1h),1.87(d,3j=8.00hz,2h)。gcqd-ab的谱图上出现了烯丙基上质子的特征吸收峰,5.16ppm和5.85ppm化学位移处的吸收峰对应于c=c上质子的吸收峰。聚合后制备的gcqd@ab-1的h1的吸收峰从3.88ppm移动到3.58ppm,说明聚合后c=c聚合成c-c,共轭程度下降,导致h1的化学位移减小,h2和h3的化学位移分别移动到1.93ppm和1.87ppm处,h2和h3分别裂分为三重峰和二重峰。以上核磁数据证实了聚合反应的完全和核壳结构的成功构建。
[0030]
图4是本实施例一制备的gcqd@ab-1甲苯溶液的荧光激发和发射光谱。gcqd@ab-1的最大激发波长是366nm,最大发射波长是418nm。在366nm光激发下,gcqd@ab-1发出强的蓝紫色荧光。gcqd@ab-1的荧光激发峰和发射峰均较窄,荧光颜色较为鲜亮。与gcqds的荧光相比,在gcqds粒子表面形成一层有机聚合物壳层后,聚合反应使c=c变成c-c,gcqds粒子表面的功能基团的共轭结构减小,导致其发光颜色蓝移,从荧光性能方面也证实了gcqds粒子表面有机聚合物壳层的形成。
[0031]
本实施方法以gcqds为荧光核,通过gcqds表面-nh2基团与烯丙基溴反应,在gcqds核粒子的表面修饰上烯丙基基团,通过自由基聚合反应成功地在gcqds核粒子的表面形成了厚度约为1nm的有机聚合物壳层,保护了gcqds核表面的缺陷能级结构。合成的核壳结构的gcqd@ab-1在空气中的荧光性能稳定,如图2所示。
[0032]
实施例二:
[0033]
本实施例二与实施例一的实验方法基本相同,参见合成路线1合成核壳结构的碳量子点gcqd@pcs,包括以下步骤:
[0034]
a.绿光碳量子点gcqds的合成参考实施例一的方法。
[0035]
b.将gcqds溶液分散于10~50ml异丙醇与1~3ml去离子水的混合溶液中,并将其转移至三颈烧瓶。在搅拌状态下加入6~12mmol k2co3和5mg正四丁基溴化铵,通氮气30~60分钟。用注射器少量多次注入4~12mmol对氯甲基苯乙烯,在80℃下回流反应,用薄层色谱监测反应进程,直至gcqds表面的-nh2反应完全。用旋蒸仪去除上述溶液的溶剂,加入10~30ml异丙醇并超声溶解糖浆状产物(gcqd-pcs),过滤除去不溶的k2co3等固体颗粒,得到10~30mlgcqd-pcs的异丙醇溶液。gcqd-pcs:1hnmr(400mhz,cdcl3):δ7.32(m,4h),6.69(d,d,3j=20.0,3j=12.0hz,1h),5.71(d,d,3j=20.0,4j=4.0hz,1h),5.19(d,d,3j=12.0,4j=
0.8hz,1h),3.51(s,2h).1h nmr数据证实了在gcqds纳米粒子表面成功接上了对-乙烯基苯甲基基团,如合成路线1所示。
[0036]
c.5mlgcqd-pcs溶液用旋转蒸发仪去除溶剂,并将产物分散于12~30ml四氢呋喃中,加入2~5ml去离子水,超声10分钟。将溶液转移至三颈烧瓶,在搅拌状态下加入5~10mg聚乙烯吡咯烷酮,5mg偶氮二异丁腈,5mg正四丁基溴化铵。通氮气30分钟后,少量多次加入用四氢呋喃稀释的2~6mmol苯乙烯溶液。在64℃下反应24h。加入5mg 2,6-二叔丁基对甲酚终止聚合反应。最终得到核壳结构的碳量子点溶液gcqd@pcs。
[0037]
实验测试分析:将本实施例制备的核壳结构的gcqd@pcs溶液作为试验样品,进行分析测试和性质检验。
[0038]
图5是实施例二制备的gcqd@pcs的高分辨透射电子显微镜图,纳米粒子均匀分散,且粒径约为3~5nm,每个gcqd@pcs碳量子点表面形成约1nm厚的有机聚合物壳层,表现出了gcqd@有机聚合物的核@壳结构。图5中的插图为局部放大2.7倍的hrtem电镜图,黑色近球形的是gcqd@pcs的核,外层白色的圆环是有机聚合物壳层。
[0039]
gcqd@pcs的荧光性能在空气中的稳定性实验。将gcqds溶液和本实施例二制备的gcqd@pcs溶液用毛细管点到薄层色谱板上,置于空气中自然放置。图2是在365nm紫外灯照射下拍摄的置于空气中不同时间下的薄层色谱的图像。gcqds在空气中,荧光信号逐渐减弱,5小时后其荧光信号基本消失。gcqd@pcs在空气中放置12天(293小时),其荧光强度基本不衰减,说明核壳结构的gcqd@pcs在空气中的稳定性远远高于gcqds,比gcqd@ab-1的荧光性能的稳定性也好。
[0040]
图6是本实施例二制备的gcqd@pcs甲苯溶液的荧光激发和发射光谱。gcqd@pcs的最大激发波长是368nm,最大发射波长是421nm。在368nm光激发下,gcqd@pcs发出强的蓝紫色荧光。与gcqds的激发峰和发射峰相比。gcqd@pcs的荧光激发峰和发射峰均明显变窄,色纯度提高,荧光的颜色较为鲜亮。与gcqds的荧光光谱相比,在gcqds粒子表面形成一层有机聚合物壳层后,聚合反应使c=c变成c-c,gcqds粒子表面的功能基团的共轭结构减小,导致其发光颜色蓝移,从荧光性能方面也证实gcqds粒子表面有机聚合物壳层的形成。
[0041]
本实施例二以gcqds为荧光核,通过gcqds表面-nh2基团与氯甲基苯乙烯反应,在gcqds核粒子的表面修饰上对乙烯苯甲基基团,通过自由基聚合反应成功地在gcqds核粒子的表面形成了厚度约为1nm的有机聚合物壳层,保护了gcqds核表面的缺陷能级结构。合成的核壳结构的gcqd@pcs在空气中的荧光性能稳定,如图2所示。
[0042]
实施例三:
[0043]
a.dad单体的合成的参见合成路线2。在三颈烧瓶中将5~10mmol亚氨基二乙酸二乙酯溶于20~60ml乙腈,加入20~40mmol k2co3,电磁搅拌。在n2气氛下逐渐升温至90℃,在搅拌状态下回流反应2-3小时。然后降温至20℃,逐滴加入5~10mmol烯丙基溴的乙腈溶液,搅拌30~60分钟后,升温至40℃加热反应,用薄层色谱监测反应进程,直至反应完全。过滤除去k2co3,滤液浓缩后用硅胶柱纯化dad单体,展开剂用乙酸乙酯和石油醚(体积比为20:1)。dad单体的1h nmr(500mhz,cdcl3):δ5.78(m,3j=6.65hz,1h),5.12(d,d,t,2j=17.19hz,3j=3.25hz,4j=1.35hz,1h),5.06(d,d,t,2j=9.95hz,3j=1.75hz,4j=0.95hz,1h),4.07(q,3j=7.2hz,4h),3.45(s,4h),3.29(d,t,3j=6.60hz,4j=1.20hz,2h),1.17(t,3j=7.1hz,6h)。1hnmr数据证实了dad的合成。
[0044]
b.gcqd-ab的合成步骤参考实施例一的a、b步骤。
[0045]
c.gcqd@ab-2的合成。典型的实验步骤如下。取5mlgcqd-ab溶液,用旋转蒸发仪去除溶剂,并将产物分散于15~25ml四氢呋喃中,加入2~5ml去离子水,超声10分钟。将溶液转移至三颈烧瓶,在搅拌状态下加入5mg聚乙烯吡咯烷酮,5mg偶氮二异丁腈,5mg正四丁基溴化铵。通氮气30分钟后,加入3~6mmol甲基丙烯酸甲酯,在64℃下加热回流反应。12小时后再加入0.6mmol dad单体继续反应12小时。加入5mg 2,6-二叔丁基对甲酚结束聚合反应。用旋转蒸发仪去除溶剂,产物重新分散于20~40ml乙醇中,逐滴加入1.8ml的2mol/l的naoh溶液,常温水解反应2小时,然后逐滴加入1.8ml的2mol/l的hcl溶液调至酸性,最终得到核壳结构的碳量子点溶液gcqd@ab-2。
[0046]
gcqd@ab-2的红外吸收光谱如图7所示,强且宽的位于2500~3500cm-1
的吸收峰是-coo-h的伸缩振动吸收峰,ν
as,-coo-(1655cm-1
),ν
s,-coo-(1387cm-1
),ν
as,-ch2-(2976cm-1
),ν
s,-ch2-(2895cm-1
)。红外光谱证实了gcqd@ab-2的合成。
[0047]
图8是本实施例三制备的gcqd@ab-2异丙醇溶液的荧光激发和发射光谱。gcqd@ab-2的最大激发波长是396nm,最大发射波长是509nm。在396nm光激发下,gcqd@ab-2发出强的蓝绿色荧光。gcqd@ab-2的荧光的strokes位移较大,113nm,gcqd@ab-2纳米粒子的浓度猝灭效应较小。
[0048]
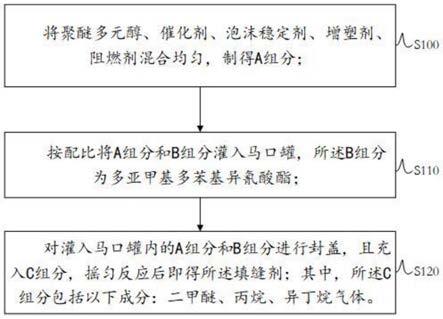
本实施方法三采用合成路线二合成了含有c=c的多羧酸有机配体dad,通过自由基聚合反应成功地将dad共聚到gcqd@ab纳米粒子的表面,合成了可以抓取ti
4
等金属离子的gcqd@ab-2荧光纳米粒子。ti
4
等金属离子可以特异性识别磷酸化蛋白,因此gcqd@ab-2荧光纳米粒子用于识别磷酸化蛋白的荧光显色试剂。
[0049]
综合上述实施例可知,本发明所制备碳量子点的特点为:
[0050]
1.通过在gcqds纳米粒子表面聚合形成厚度约为1nm有机聚合物壳层,保护了gcqds纳米粒子表面缺陷能级结构,大大地提高了其在空气中的荧光性能的稳定性。
[0051]
2.gcqds纳米粒子表面的有机聚合物壳层降低了gcqds的表面能,粒子在溶液中的分散性更好,在溶液中可以长期保存。
[0052]
3.gcqd@ab-1、gcqd@pcs和gcqd@ab-2均可以用紫外光激发,发出明亮的蓝紫色荧光或蓝绿荧光,用于荧光显色试剂可以避免激发光的干扰。
[0053]
4.本发明可以通过简单的自由基共聚反应连接其它可识别生物分子或离子的功能基团。
[0054]
5.本发明实验方法合成条件简单,不使用复杂的设备,合成成本较低。
[0055]
6.本发明制备的碳量子点荧光纳米粒子的荧光性能在空气中相当稳定,可用于固相检测条件下的荧光显色试剂。如通过gcqd@ab-2荧光纳米粒子抓取ti
4
等金属离子的,ti
4
等金属离子可以特异性识别磷酸化蛋白,用于磷酸化蛋白的荧光显色试剂。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。