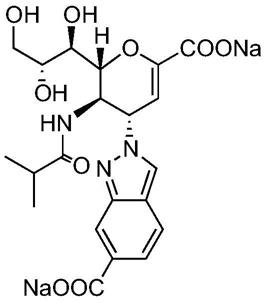
1.本发明涉及医学治疗领域。更特别地,本发明涉及新型抗病毒剂和它们在治疗由病毒感染引起的疾病或病况中的用途。
背景技术:
2.本文对背景技术的任何引用不应被解释为承认此类技术构成澳大利亚或其他地方的公知常识。
3.病毒导致一系列给社会构成巨大代价的哺乳动物疾病。病毒感染的影响范围可从常见的流感症状到严重的呼吸问题,并可能导致死亡,特别是在社区的年轻人、老年人和免疫功能低下的成员中。
4.正粘病毒科(orthomyxoviridae)病毒(包括甲型、乙型和丙型流感病毒)和副粘病毒科(paramyxoviridae)病毒是每年造成大量人类感染的病原生物体。
5.以副粘病毒科为例,人副流感病毒1型和3型(hpiv-1和3)是婴儿和幼儿上呼吸道和下呼吸道疾病的主要原因,并且影响老年人和免疫功能低下者。值得注意的是,据估计,仅在美国,每年就有高达500万例下呼吸道感染发生在5岁以下的儿童中,并且在这些病例中有大约三分之一已分离出hpiv。移植患者中经常报告hpiv感染,造血干细胞移植患者的死亡率高达30%。尽管做出了持续的努力,但目前既没有疫苗也没有特定的抗病毒疗法来分别预防或治疗hpiv感染。一些最近的方法集中于进入阻断和通过小分子触发过早的病毒融合。
6.副流感病毒与宿主细胞的初始相互作用是通过其表面糖蛋白、血细胞凝集素-神经氨酸酶(hn)并且涉及识别含n-乙酰神经氨酸的糖缀合物。副流感病毒hn是一种多功能蛋白,其涵盖不仅在一种蛋白质内,而且显然在单个结合位点中的受体结合(用于细胞粘附)和受体破坏(促进病毒释放)的功能。此外,hn参与启动靶宿主细胞感染所需的病毒表面融合(f)蛋白的活化。因此血细胞凝集素-神经氨酸酶的抑制可提供用于抗病毒剂的靶标。
7.某些抗病毒化合物已在本技术人较早提交的国际申请(公布为wo2016/033660)中公开为病毒血细胞凝集素-神经氨酸酶功能的调节剂。尽管适合其目的,但该公开在某些关键位置可耐受的可变性和功效的最佳替代方面提供了有限的指导。
技术实现要素:
8.根据本发明的第一方面,提供式(i)化合物或其药学上可接受的盐:
[0009][0010]
其中r1选自由cooh或其盐、c(o)nr9r
10
、c(o)or
11
组成的组,其中r9、r
10
和r
11
独立地选自由氢、任选取代的c
1-c6烷基和任选取代的芳基组成的组;
[0011]
r3选自由以下组成的组:任选取代的n-连接的萘并三唑、任选取代的n-连接的吲唑和下式的n-连接的三唑:
[0012][0013]
其中r
20
选自由以下组成的组:
[0014]
其中*是连接点,并且r
21
、r
22
和r
23
独立地选自由以下组成的组:任选取代的烷基、任选取代的芳基、任选取代的烷基杂环基、任选取代的烷基杂芳基、任选取代的烷基胺基、任选取代的二烷基胺基和任选取代的接头,所述接头将所述化合物连接到另一式(i)化合物;
[0015]
r4选自由磺酰胺基、脲基和nhc(o)r
17
组成的组,其中r
17
选自由c
1-c6烷基、c
1-c6卤代烷基和c
3-c6环烷基组成的组,所述基团均可任选地被取代;
[0016]
r6、r7和r8独立地选自由以下组成的组:oh、受保护的oh、nh2、c
1-c6烷基、c
1-c6卤代烷基、nr
18r18’、c
1-c6烷氧基、c
1-c6卤代烷氧基、-oc(o)r
18
、-nh(c=o)r
18
和s(o)
nr18
,其中n=0-2并且每个r
18
和r
18’均在适当时独立地选自氢、任选取代的c
1-c6烷基和任选取代的c
1-c9烷酰基。
[0017]
在第一方面的一个实施方案中,式(i)化合物是式(ii)化合物:
[0018][0019]
其中r1、r3、r4、r6、r7和r8如上文所述。
pocket)的新化合物模板。本文所公开的某些化合物还将抑制剂支架延伸到hpiv hn结合袋之外,以获得有益的结合相互作用的增益,从而改善效力和多价性。
[0037]
定义
[0038]
在本专利说明书中,术语“包含(comprises/comprising)”、“包括(includes/including)”或类似术语意在意非排他性包括,使得包含要素列表的方法或组合物不仅包括这些要素,而且还可能包括未列出的其他元素。
[0039]
除非另外定义,否则本文使用的所有技术术语和科学术语的含义都与本发明所属领域的普通技术人员通常所理解的含义相同。
[0040]
如本文所用,“有效量”是指相关活性剂足以预防所治疗病况的症状的发生、或使症状恶化停止或治疗并缓解或至少减少症状的严重程度的施用量。有效量将以本领域技术人员理解的方式随患者年龄、性别、体重等而变化。适当的剂量或给药方案可通过常规试验确定。
[0041]
如本文所用的术语“药学上可接受的盐”是指对于全身或局部施用在毒理学上安全的盐,诸如由药学上可接受的无毒碱或无毒酸(包括无机碱或有机碱,和无机碱或有机碱)制备的盐。药学上可接受的盐可选自包括以下的组:碱盐和碱土金属盐、铵盐、铝盐、铁盐、胺盐、葡糖胺盐、氯化物盐、硫酸盐、磺酸盐、亚硫酸氢盐、硝酸盐、柠檬酸盐、酒石酸盐、酒石酸盐、酒石酸氢盐(bitarate)、磷酸盐、碳酸盐、碳酸氢盐、苹果酸盐、马来酸盐、萘磺酸盐、富马酸盐、琥珀酸盐、乙酸盐、苯甲酸盐、对苯二甲酸盐、棕榈酸盐、哌嗪盐、果胶酸盐(pectinate)和s-甲基甲硫氨酸盐等。
[0042]
术语“取代的”和“任选取代的”在其在本文中每次使用时,以及在没有明确列出任何特定部分的情况下,是指相关部分(例如烷基链或环结构)被一个或多个选自以下的基团取代:c
1-c6烷基、c
1-c6卤代烷基、c
1-c6烷氧基、c
1-c6卤代烷氧基(诸如三氟甲氧基、三氟乙氧基等)cn、oh、氧代、nh2、nr
28r28’(其中r
28
和r
28’独立地选自氢、任选取代的c
1-c9烷基、任选取代的芳基、r
29
c=o、r
29
so2和r
29
nhc=o,其中r
29
是c
1-c9烷基)、cl、f、br、i、芳基和杂环基,所述后两个部分本身可任选地被取代。当该术语在叙述多个官能团之前使用时,则除非另外显而易见,否则其意在适用于所有列出的官能团。例如,“任选取代的氨基、杂环、芳基”意指氨基、杂环和芳基均可任选地被取代。在其中相关基团是r3并且其连接到另一式(i)化合物以形成二聚体的实施方案中,则所述部分(例如“任选取代的烷基”或“任选取代的烷基杂芳基/烷基杂环基”)可被包含烷基链和/或三唑环的接头取代,所述部分通过所述接头连接到另一式(i)化合物的r3,形成所述二聚体。
[0043]
术语“烷基”是指含有例如1到约12个碳原子、优选1到约8个碳原子、更优选1到约6个碳原子、甚至更优选1到约4个碳原子、还更优选1到2碳原子的直链或支链烷基取代基。此类取代基的示例包括甲基、乙基、丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、戊基、异戊基、2-甲基丁基、3-甲基丁基、己基、庚基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2-乙基丁基、3-乙基丁基、辛基、壬基、癸基、十一烷基、十二烷基等。所指的碳数涉及碳主链和碳分支,但不包括属于任何取代基的碳原子,例如从主碳链分支的烷氧基取代基的碳原子。
[0044]
术语“环烷基”是指任选取代的非芳族单环、二环或三环碳基团。在适当情况下,环烷基可具有指定数目的碳原子,例如,c
3-c6环烷基是具有3、4、5或6个碳原子的碳环基团。非限制性示例可包括环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基等。在
一些实施方案中,“环烷基”是指任选取代的饱和单环、二环或三环碳基团。
[0045]
术语“芳基”是指如本领域通常所理解的未取代或取代的芳族碳环取代基。应当理解,根据休克尔规则(h
ü
ckel’s rule),术语芳基适用于为平面且包含4n 2个π电子的环状取代基。优选c-6芳基。
[0046]
如本文所用的术语“杂环”和“杂环基”具体涉及某些“r”基团时是指通过从杂环化合物的环原子中去除氢原子而获得的部分,所述杂环化合物在环中可具有5到7个原子,并且在这些原子中有1到4个是杂原子,所述环与第二环分离或稠合,其中所述杂原子独立地选自o、n和s。杂环和杂环基包括芳族杂环基和非芳族杂环基。杂环体系可经由基团的任何数目的碳原子或杂原子连接到另一部分,并且均可以是饱和的和不饱和的。杂环体系可经由基团的任何数目的碳原子或杂原子连接到另一部分,并且均可以是饱和的和不饱和的。杂环基的非限制性示例可选自吡唑基、咪唑基、吲哚基、异吲哚基、三唑基、苯并三唑基、四唑基、嘧啶基、吡啶基、吡嗪基、二嗪基、三嗪基、四嗪基、吡咯烷基、吡咯啉基、吡喃基、哌啶基、哌嗪基、吗啉基、四氢呋喃基、四氢噻吩基、吡唑啉基、二硫杂环戊二烯基(dithiolyl)、氧杂硫杂环戊二烯基(oxathiolyl)、二烷基、二氧杂环己烯基(dioxinyl)、嗪基、氮杂环庚三烯基(azepinyl)、二氮杂环庚三烯基(diazepinyl)、硫杂氮杂环庚三烯基(thiazepinyl)、氧杂环庚三烯基(oxepinyl)和硫杂环庚三烯基(thiapinyl)、咪唑啉基、硫代吗啉基等。
[0047]
术语“杂芳基”或“芳族杂环基”是指含有一个或多个(特别是一到四个)非碳原子(特别是n、o或s)或其组合的芳基,所述杂芳基任选地在一个或多个碳原子或氮原子处被取代。杂芳基环还可与一个或多个环状烃、杂环、芳基环或杂芳基环稠合。杂芳基包括但不限于具有一个杂原子的5元杂芳基(例如,噻吩基、吡咯基、呋喃基);在1,2位或1,3位具有两个杂原子的5元杂芳基(例如,唑基、吡唑基、咪唑基、噻唑基、嘌呤基);具有三个杂原子的5元杂芳基(例如,三唑基、噻二唑基);具有四个杂原子的5元杂芳基(例如,四唑基);具有一个杂原子的6元杂芳基(例如,吡啶基、喹啉基、异喹啉基、菲基、5,6-环庚烯并吡啶基);具有两个杂原子的6元杂芳基(例如,哒嗪基、噌啉基、酞嗪基、吡嗪基、嘧啶基、喹唑啉基);具有三个杂原子的6元杂芳基(例如,1,3,5-三嗪基);和具有四个杂原子的6元杂芳基。“取代的杂芳基”意指具有一个或多个非干扰基团作为取代基且包括在
‘
任选取代的’下所定义的那些的杂芳基。杂芳基的示例包括噻吩基、苯并噻吩基、苯并呋喃基、苯并咪唑基、苯并唑基、苯并噻唑基、苯并异噻唑基、萘并[2,3-b]噻吩基、呋喃基、异吲嗪基(isoindolizine)、二苯并哌喃(xantholene)、吩嗪基(phenoxatine)、吡咯基、咪唑基、吡唑基、吡啶基、吡嗪基、嘧啶基、哒嗪基、吲哚基、异吲哚基、1h-吲唑基、嘌呤基、喹啉基、异喹啉基、酞嗪基、萘啶基、喹喔啉基、噌啉基(cinnoline)、咔唑基、菲啶基、吖啶基、吩嗪基、噻唑基、异噻唑基、吩噻嗪基、唑基、异唑基、呋咱基(furazane)、吩嗪基、2-吡啶基、3-吡啶基或4-吡啶基、2-喹啉基、3-喹啉基、4-喹啉基、5-喹啉基或8-喹啉基、1-异喹啉基、3-异喹啉基、4-异喹啉基或5-异喹啉基、1-吲哚基、2-吲哚基或3-吲哚基和2-噻吩基或3-噻吩基。所述基团可以是端基或桥连基。
[0048]
术语“烷基胺基”和“二烷基胺基”分别是指-nhr和-nrr’基团,其中,“r”和“r
’”
是烷基,其是任选取代的,并且可独立地如上文所定义。也就是说,r和r’可以是但不一定是相
同的烷基部分。
[0049]
术语“胺基”可以是指如上文所定义的-nh2、“烷基胺基”和“二烷基胺基”。
[0050]
术语“受保护的oh”或“受保护的羟基”是指用常见保护基团保护的羟基,所述保护基团诸如酰基、醚基或酯基,包括c
1-c3酰基、用于形成醚基的c
1-c4烷基或形成醚基的芳基(诸如苄基)或c
1-c4酯基。
[0051]
如本文所用的术语“n-连接的”关于第一方面的化合物(包括式(i)和(ii)的化合物,例如“n-连接的三唑”、“n-连接的萘并三唑”、“n-连接的吲唑”或“n-连接的杂环”),是指连接在神经氨酸核心的c4位的部分(式(i)和(ii)中的r3),并将该连接限制为涉及环碳和氮原子之间的直接连接。优选地,它是指r3部分经由氮原子连接到神经氨酸核,该氮原子本身形成适当杂环的一部分,诸如三唑环、吲唑、萘并三唑等的一个氮。
[0052]
每当指出结构中的原子数范围(例如,c
1-c
12
、c
1-c
10
、c
1-c9、c
1-c6、c
1-c4烷基等)时,特别预期还可使用落入所示范围内的任何子范围或单个碳原子数。因此,例如,如关于本文所提到的任何化学基团(例如,烷基等)所用的1-12个碳原子(例如,c
1-c
12
)、1-9个碳原子(例如,c
1-c9)、1-6个碳原子(例如,c
1-c6)、1-4个碳原子(例如,c
1-c4)、1-3个碳原子(例如,c
1-c3)或2-8个碳原子(例如,c
2-c8)的范围的叙述涵盖且特别描述视情况1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个和/或12个碳原子,以及其任何子范围(例如,视情况1-2个碳原子、1-3个碳原子、1-4个碳原子、1-5个碳原子、1-6个碳原子、1-7个碳原子、1-8个碳原子、1-9个碳原子、1-10个碳原子、1-11个碳原子、1-12个碳原子、2-3个碳原子、2-4个碳原子、2-5个碳原子、2-6个碳原子、2-7个碳原子、2-8个碳原子、2-9个碳原子、2-10个碳原子、2-11个碳原子、2-12个碳原子、3-4个碳原子、3-5个碳原子、3-6个碳原子、3-7个碳原子、3-8个碳原子、3-9个碳原子、3-10个碳原子、3-11个碳原子、3-12个碳原子、4-5个碳原子、4-6个碳原子、4-7个碳原子、4-8个碳原子、4-9个碳原子、4-10个碳原子、4-11个碳原子和/或4-12个碳原子等)。
[0053]
如本文所用,术语“受试者”或“个体”或“患者”可指期望疗法的任何受试者,特别是脊椎动物受试者,且甚至更特别是哺乳动物受试者。适合的脊椎动物包括但不限于灵长类动物、鸟类、家畜动物(例如,绵羊、牛、马、驴、猪)、实验室测试动物(例如,兔、小鼠、大鼠、豚鼠、仓鼠)、伴侣动物(例如,猫、狗)和捕获的野生动物(例如,狐狸、鹿、澳洲野犬)。优选的受试者是需要治疗由病毒感染引起的疾病或病况的人。然而,应当理解,上述术语并不意味着一定存在症状。
[0054]
本文提及的“血细胞凝集素-神经氨酸酶”、“血细胞凝集素-神经氨酸酶蛋白”等可被认为可与“血细胞凝集素和/或神经氨酸酶功能”互换。它们可被认为并入阻断血细胞凝集功能或抑制神经氨酸酶(酶)功能中的一种或多种。因此血细胞凝集功能的阻断可涉及血细胞凝集素-神经氨酸酶蛋白的调节、阻断或抑制,不希望受任何理论束缚,这可能是本文所述化合物的一种作用机制。
[0055]
根据本发明的第一方面,提供式(i)化合物或其药学上可接受的盐:
[0056][0057]
其中r1选自由cooh或其盐、c(o)nr9r
10
、c(o)or
11
组成的组,其中r9、r
10
和r
11
独立地选自由氢、任选取代的c
1-c6烷基和任选取代的芳基组成的组;
[0058]
r3选自由以下组成的组:任选取代的n-连接的萘并三唑、任选取代的n-连接的吲唑和下式的n-连接的三唑:
[0059][0060]
其中r
20
选自由以下组成的组:
[0061]
其中*是连接点,r
21
、r
22
和r
23
独立地选自由以下组成的组:任选取代的烷基、任选取代的烯基、取代的炔基、任选取代的环烷基、任选取代的芳基、任选取代的杂芳基、任选取代的杂环基、任选取代的烷基杂环基、任选取代的烷基杂芳基、任选取代的烷基胺基、任选取代的二烷基胺基和任选取代的接头,所述接头将所述化合物连接到另一式(i)化合物;
[0062]
r4选自由磺酰胺基、脲基和nhc(o)r
17
组成的组,其中r
17
选自由c
1-c6烷基、c
1-c6卤代烷基和c
3-c6环烷基组成的组,所述基团均可任选地被取代;
[0063]
r6、r7和r8独立地选自由以下组成的组:oh、受保护的oh、nh2、c
1-c6烷基、c
1-c6卤代烷基、nr
18r18’、c
1-c6烷氧基、c
1-c6卤代烷氧基、-oc(o)r
18
、-nh(c=o)r
18
和s(o)
nr18
,其中n=0-2并且每个r
18
和r
18’均在适当时独立地选自氢、任选取代的c
1-c6烷基和任选取代的c
1-c9烷酰基。
[0064]
在第一方面的一个实施方案中,式(i)化合物是式(ii)化合物:
[0065][0066]
其中r1、r3、r4、r6、r7和r8如前文所述。
[0067]
在式(i)或(ii)化合物的一个实施方案中,r1是cooh或其盐或c(o)or
11
,其中r
11
选自甲基、乙基和丙基。
[0068]
在某些具体实施方案中,r1选自由cooh、coona和c(o)ome组成的组。
[0069]
在式(i)或(ii)化合物的一个实施方案中,当r3是任选取代的n-连接的萘并三唑时,其具有下式:
[0070][0071]
其中ra、rb、rc、rd、re和rf独立地选自由以下组成的组:氢、羟基、氰基、卤基、酰氨基、c
1-c
12
烷基、c
1-c
12
烷氧基、c
1-c
12
卤代烷氧基、c
1-c
12
烷酰基、c
1-c
12
卤代烷酰基、c
1-c
12
卤代烷基、吡啶基和苯基,所述基团均可视情况任选地被取代。
[0072]
在某些实施方案中,ra、rb、rc、rd、re和rf独立地选自由以下组成的组:氢、羟基、氰基、卤基、乙酰氨基、c
1-c6烷基、c
1-c9烷氧基、c
1-c9烷酰基、c
1-c6卤代烷基、任选取代的吡啶基和任选取代的苯基。
[0073]
在某些实施方案中,ra和rb;rb和rc;rc和rd;rd和re;以及re和rf中的一个或多个可形成5或6元芳基或杂芳基或杂环。
[0074]
在式(i)或(ii)化合物的一个实施方案中,当r3是任选取代的n-连接的吲唑时,其具有下式:
[0075][0076]
其中rg、rh、ri和rj独立地选自由以下组成的组:氢、羟基、卤基、c
1-c6烷氧基、c
1-c6卤代烷基、氰基、磺酰基、胺基、酰氨基和羧基;并且
[0077]
rg和rh、rh和ri以及ri和rj可一起形成杂芳基环、杂环或芳基环,所述环中的每一个均可任选地被取代。
[0078]
在实施方案中,rg和rh、rh和ri以及ri和rj可一起形成5元、6元或7元的杂芳基环、杂环或芳基环(尤其是5或6元杂芳基环、杂环或芳基环;更尤其是1,3-二氧戊环、吡啶环、噻吩环、咪唑环、吡咯环或苯基环),所述环中的每一个均可任选地被取代(尤其是被卤基和氰基中的至少一种取代;更尤其是被f、br和氰基中的至少一种取代)。
[0079]
在某些实施方案中,rg选自由以下组成的组:氢、羟基、氰基、卤基(包括氟)、c
1-c6烷氧基、酰氨基和羧基。
[0080]
在实施方案中,rh选自由以下组成的组:氢、羟基、卤基(包括氟或溴)、氰基、c
1-c6卤代烷基、c
1-c6烷氧基、磺酰基、羧基和胺基。
烷基-氮杂芳基、任选取代的c
1-c
12
烷基胺基、任选取代的c
1-c
12
二烷基胺基、任选取代的c
1-c6烷基-nh-co-芳基、任选取代的c
1-c6烷基-nh-co-芳基-芳基、任选取代的c
1-c6烷基-nh-co-环烷基、任选取代的c
1-c6烷基-nh-so
2-芳基、任选取代的c
1-c6烷基-nh-so
2-c
1-c6烷基-芳基和任选取代的接头,所述接头将所述化合物连接到另一式(i)化合物。
[0098]
在实施方案中,r
21
、r
22
和r
23
独立地选自由以下组成的组:任选取代的c
1-c
12
烷基、任选取代的c
2-c
12
烯基、任选取代的c
2-c
12
炔基、任选取代的c6环烷基、任选取代的5或6元芳基、任选取代的5或6元杂芳基、任选取代的5或6元杂环基、任选取代的c
1-c9烷基5或6元杂环、任选取代的c
1-c9烷基5或6元杂芳基、任选取代的c
1-c
12
烷基胺基、任选取代的c
1-c
12
二烷基胺基、任选取代的c
1-c6烷基-nh-co-芳基、任选取代的c
1-c6烷基-nh-co-芳基-芳基、任选取代的c
1-c6烷基-nh-co-环烷基、任选取代的c
1-c6烷基-nh-so
2-芳基、任选取代的c
1-c6烷基-nh-so
2-c
1-c6烷基-芳基和任选取代的接头,所述接头将所述化合物连接到另一式(i)化合物。
[0099]
在实施方案中,r
21
、r
22
和r
23
独立地选自由以下组成的组:任选取代的c
1-c9烷基、任选取代的c
2-c9烯基、任选取代的c
2-c9炔基、任选取代的c6环烷基、任选取代的5或6元芳基、任选取代的5或6元氮杂芳基、任选取代的5或6元氮杂环基、任选取代的c
1-c6烷基5或6元杂环、任选取代的c
1-c6烷基5或6元氮杂芳基、任选取代的c
1-c9烷基胺基、任选取代的c
1-c9二烷基胺基、任选取代的c
1-c6烷基-nh-co-苯基、任选取代的c
1-c6烷基-nh-co-苯基-苯基、任选取代的c
1-c6烷基-nh-co-(3到6元)环烷基、任选取代的c
1-c6烷基-nh-so
2-苯基、任选取代的c
1-c6烷基-nh-so
2-c
1-c6烷基-苯基和任选取代的接头,所述接头将所述化合物连接到另一式(i)化合物。
[0100]
在特定实施方案,r
21
可选自任选取代的c
1-c9烷基、c
2-c9烯基和c
2-c9炔基,其中相关链的末端碳连接到选自由以下组成的组的部分:叠氮基、任选取代的氨基和任选取代的5元氮杂芳基。优选地,任选取代的5元氮杂芳基选自由以下组成的组:吡咯基、咪唑基、吡唑基、三唑基、四唑基、苯并三唑基和异吲哚基,所述基团中的每一个可视情况任选地被取代。更优选地,任选取代的5元氮杂芳基是任选取代的三唑。
[0101]
在特定实施方案中,r
21
可选自任选取代的c
1-c9烷基、c
2-c9烯基、c
2-c9炔基、任选取代的c
1-c6烷基氨基、任选取代的苯基(其中任选的取代基可尤其选自由以下组成的组中的至少一种:卤基、-oc
1-c6烷基、o-c
1-c
6-卤代烷基、硝基和c
1-c
6-烷基)、任选取代的c
1-c6烷基-nhco-苯基(其中苯基可尤其任选地被由以下组成的组中的至少一种取代:-n(c
1-c6烷基)2和苯基)、任选取代的c
1-c6烷基-nhso
2-苯基(其中苯基可尤其任选地被硝基取代)、任选取代的c
1-c6烷基-nhso
2-c
1-c6烷基-苯基和任选取代的c
1-c6烷基-nhco-(3到6元)环烷基。
[0102]
在特定实施方案,r
23
可选自c
1-c6烷基和任选取代的苯基(其中任选的取代基可尤其是硝基)。
[0103]
当r
21
是将化合物连接到另一式(i)化合物的任选取代的接头时,则所述接头可选自任选取代的c
1-c
12
烷基;任选取代的c
1-c9烷基;任选取代的c
2-c9烯基;和任选取代的c
2-c9炔基;所述基团中的任一种可连接到5元氮杂芳基。合适地,5元氮杂芳基可以是三唑。
[0104]
当r
21
是将化合物连接到另一式(i)化合物的任选取代的接头时,则式(i)化合物可具有下式:
[0105]
接头
[0106]
其中r1、r4、r6、r7和r8如前文所述并且接头选自c
1-c
12
烷基;c
1-c9烷基;c
2-c9烯基;和c
2-c9炔基;所述基团中的任一个可任选地被取代并且任选地连接到5元氮杂芳基。
[0107]
任选的取代基可如前文所定义,特别优选羟基、芳基、杂芳基、酰氨基和醚基中的一个或多个。
[0108]
在某些实施方案中,接头选自以下:
[0109]c1-c
12
烷基、c
1-c9烷基、c
2-c9烯基、c
2-c9炔基、
[0110][0111]
并且其中所提到的c
1-c
12
烷基、c
1-c6烷基和c
1-c
20
烷基部分均可任选地被羟基、芳基、杂芳基、酰氨基和醚基中的一个或多个取代。
[0112]
在具有连接两个三唑环的c1到c
20
烷基的以上结构中,c1到c
20
烷基可在实施方案中选自由以下组成的组:c1到c
16
烷基、c1到c
12
烷基、c1到c9烷基和c1到c6烷基,所述烷基团均可任选地被芳基、杂芳基、酰氨基和醚基中的一个或多个取代。
[0113]
在某些实施方案中,r3可选自由以下组成的组:
[0114]
[0115]
[0116]
[0117][0118]
[0119]
上文所列任何r3基团的具体部分或公开内容可与如本文所述的r1、r4、r6、r7或r8基团的任何公开内容组合。
[0120]
在前述实施方案中的任一个中,r4可选自由以下组成的组:-nhs(o)2r
27
,其中r
27
选自由c
1-c6烷基、c
1-c6卤代烷基和c
3-c6环烷基组成的组,所述基团均可任选地被取代;-nhc(o)nhr
17
,其中r
17
可如前文所定义;和以下基团:
[0121][0122]
在针对第一方面所述的任一式的实施方案中,r4选自由以下组成的组:-nhac、-nhc(o)ch(ch3)2、-nhc(o)cf3和-nhc(o)ch2ch3。
[0123]
在式(i)或(ii)的化合物的任一实施方案中,r6、r7和r8可独立地选自由以下组成的组:oh、c
1-c3烷氧基、-oc(o)r
18
(其中r
18
是任选取代的c
1-c3烷基)和-nr
18r18’(其中r
18
和r
18’选自氢、任选取代的c
1-c3烷基和任选取代的c
1-c6烷酰基)。当r
18
是c(o)r(即烷酰基)时,则
‘
r’可以是c
1-c6烷基或c
5-c6环烷基。
[0124]
在针对第一方面所述的任一式的实施方案中,r6、r7和r8可独立地选自oh和oac。
[0125]
在一个实施方案中,式(i)化合物可以是式(iiia)或(iiib)化合物:
[0126][0127]
其中r1、r4、r6、r7、r8、ra、rb、rc、rd、re和rf如前文所述。
[0128]
在式(iiia)和式(iiib)的某些实施方案中,r1可选自cooh或其盐或c(o)or
11
,其中r
11
选自甲基、乙基和丙基,优选地r1选自cooh、coona和c(o)ome;r4选自由以下组成的组:-nhac、-nhc(o)ch2(ch3)2、-nhc(o)cf3、-nhc(o)ch2ch3、-nhs(o)2r
27
(其中r
27
选自由c
1-c6烷基、c
1-c6卤代烷基和c
3-c6环烷基组成的组,所述基团均可任选地被取代)、-nhc(o)nhr
17
(其中r
17
可如前文所定义);并且r6、r7和r8可独立地选自oh和oac。
[0129]
在一个实施方案中,式(i)化合物可以是式(iva)或(ivb)化合物:
[0130][0131]
其中r1、r4、r6、r7、r8、rg、rh、ri和rj如前文所述。
[0132]
在式(iva)和式(ivb)的某些实施方案中,r1可选自cooh或其盐或c(o)or
11
,其中r
11
选自甲基、乙基和丙基,优选地r1选自cooh、coona和c(o)ome;r4选自由以下组成的组:-nhac、-nhc(o)ch2(ch3)2、-nhc(o)cf3和-nhc(o)ch2ch3;并且r6、r7和r8可独立地选自oh和oac。
[0133]
在一个实施方案中,式(i)化合物可以是式va、vb、via、vib、viia和viib中任一种或多种的化合物:
[0134][0135][0136]
其中r1、r4、r6、r7、r8、r
21
、r
22
和r
23
如前文针对第一方面的任一个或多个实施方案所定义。
[0137]
在式(va)和(vb)的一个实施方案中,所述化合物具有下式:
[0138][0139]
其中n是1到9,优选1到6,并且其中r
24
选自由以下组成的组:叠氮基;5元氮杂芳基,其任选地与又一环系稠合;coor
30
,其中r
30
选自氢、c
1-c
12
烷基和芳基;和-nr
25r26
,其中r
25
和r
26
独立地选自氢和c
1-c6烷基。
[0140]
在实施方案中,当r
24
是任选地与又一环系稠合的5元氮杂芳基时,则其可与包括苯基环的5或6元芳基环稠合。例如,可形成异吲哚和类似稠环体系。
[0141]
上式化合物可适用于形成如本文所述的式(i)化合物的二聚体。也就是说,某些式va和vb化合物可用于转化为式(i)的二聚体。
[0142]
在式(i)和式(ii)的实施方案中,所述化合物可选自由以下组成的组:
[0143]
[0144]
[0145]
[0146]
[0147]
[0148]
[0149][0150]
以及其受保护形式,包括替代游离羟基处的氢的乙酰基,其所有c-2类似物,其中c-2羧基呈质子化形式、钠盐形式或前药形式并且其中可认为每种化合物均具有所公开的接近类似物,其中r4位明确地被任一-nhc(o)r基团替代,其中r是c
1-c4烷基或卤代烷基。
[0151]
合成化学领域的技术人员将了解,cooh基团容易与盐形式或酯保护基团(例如甲酯基团)互换,因此所有此类形式均被认为是本文参考上文列出的化合物公开的。
[0152]
以上化合物的前药形式可明确地认为包括c1-c20酯或包含环烷基或芳基部分的酯。芳基部分可包括取代的苯基或稠合的2-3环状芳族环。
[0153]
在一个实施方案中,第一方面的化合物是血细胞凝集素-神经氨酸酶调节剂。也就是说,第一方面的化合物是血细胞凝集素和/或神经氨酸酶功能的调节剂。优选地,第一方面的化合物是血细胞凝集素-神经氨酸酶抑制剂。也就是说,血细胞凝集素和/或神经氨酸酶功能的抑制剂。这可以包括通过调节血细胞凝集素蛋白来阻断血细胞凝集功能。
[0154]
在一个实施方案中,可能优选的是,血细胞凝集素-神经氨酸酶抑制剂是流感或副流感血细胞凝集素和/或神经氨酸酶抑制剂或阻断剂。换句话说,在一个实施方案中,可能优选的是,血细胞凝集素和/或神经氨酸酶功能抑制剂是流感或副流感血细胞凝集素和/或神经氨酸酶功能抑制剂。这可以包括阻断流感或副流感血细胞凝集功能以及因此调节流感血细胞凝集素蛋白或副流感血细胞凝集素-神经氨酸酶蛋白
[0155]
可采用许多合成途径来获得本发明的化合物。实验部分详述了合成本发明的某些抑制剂以用作参考化合物的某些途径。相关合成技术(其也可应用于第一方面的化合物的合成)公开于以下文献中:nature scientific reports,7:4507,2017年7月3日;angew.chem.int.ed.2015,54,2936-2940;nature scientific reports,6:24138,2016年4月7日;med.chem.commun.,2017,8,130-134;j.med.chem.2014,57,7613-7623;carbohydr.res.244,181-185(1993);nature communications,5:5268,2014年10月20日;
viruses,2019,11,417,2019年5月5日;carbohydr.res.342,1636-1650(2007);bioorg.med.chem.lett.16,5009-5013(2006);pct申请wo2002076971;和pct申请wo2016033660,这些文献中的每一个均在此以引用方式整体并入。此类技术和合成方法可用于获得第一方面的所有化合物。
[0156]
根据本发明的第二方面,提供一种药物组合物,其包含有效量的第一方面的作一实施方案或式的化合物或其药学上可接受的盐,以及药学上可接受的载体、稀释剂和/或赋形剂。
[0157]
合适地,所述药物组合物用于治疗或预防由病毒感染引起的疾病、病症或病况。
[0158]
所述药物组合物可包含超过一种式(i)化合物。当所述组合物包含超过一种化合物时,则所述化合物可以是任何比率。所述组合物可进一步包含已知的助活性物、递送媒介物或佐剂。
[0159]
第一方面的任一实施方案或式的化合物以足以抑制或改善作为治疗对象的疾病、病症或病况的量存在于药物组合物中。本领域技术人员可容易地确定化合物和含有此类化合物的药物组合物的适合剂型和速率。
[0160]
剂型可包括片剂、分散剂、合剂、气溶胶、悬浮液、注射剂、溶液、糖浆、口含片、胶囊等。
[0161]
本发明的第三方面在于治疗患者的由病毒感染引起的疾病、病症或病况的方法,其包括向所述患者施用有效量的第一方面的任一实施方案或式的化合物或其药学有效盐或第二方面的药物组合物的步骤。
[0162]
本发明的第四方面提供第一方面的任一实施方案或式的化合物或其药学有效盐或第二方面的药物组合物,其用于治疗患者的由病毒感染引起的疾病、病症或病况。
[0163]
本发明的第五方面提供第一方面的任一实施方案或式的化合物或其药学有效盐在制造用于治疗由病毒感染引起的疾病、病症或病况的药剂中的用途。
[0164]
在第三、第四或第五方面的一个实施方案中,疾病、病症或病况是由流感和/或副流感病毒引起的感染。
[0165]
感染可由以下中的一种或多种引起:甲型流感病毒、乙型流感病毒、丙型流感病毒、丁型流感病毒、副流感病毒、呼吸道合胞病毒(rsv)和人偏肺病毒(hmpv)。
[0166]
当疾病、病症或病况为副流感病毒感染时,其可选自由hpiv-1、hpiv-2、hpiv-3和hpiv-4病毒组成的组。这些可包括所有病毒亚型,例如4a和4b。
[0167]
当疾病、病症或病况由rsv引起时,则其可以是a和/或b亚型,例如,hrsv-a和hrsv-b。
[0168]
当疾病、病症或病况由hmpv引起时,则其可由hmpv a1、a2、b1和b2亚型中的任一种或多种引起。
[0169]
优选地,患者是驯养动物或家畜动物或人。
[0170]
本发明的第六方面提供调节病毒血细胞凝集素和/或神经氨酸酶功能的方法,其包括使病毒血细胞凝集素-神经氨酸酶与第一方面的任一实施方案或式的化合物接触的步骤。
[0171]
优选地,调节涉及抑制病毒血细胞凝集素和/或神经氨酸酶功能或病毒血细胞凝集素-神经氨酸酶。
[0172]
以下实验部分更详细地描述本发明的某些化合物和它们的抗病毒活性的表征。意在说明本发明化合物的某些具体实施方案和它们的功效,而不以任何方式限制本发明。
[0173]
实验
[0174]
化学
[0175]
一般方法
[0176]
试剂和无水溶剂购自商业来源并且不进一步纯化即使用。在烘干的玻璃器皿中,在氩气气氛下进行无水反应。使用薄层色谱(tlc)在预先涂覆有硅胶60f254(e.merck)的铝板上监测反应。在254nm的紫外光下观察显色的板,然后使其在施加h2so4于etoh中的溶液(5%v/v)、之后炭化后显现。在硅胶60(0.040-0.063mm)上使用蒸馏溶剂进行快速色谱。在brukeravance 400mhz光谱仪上分别在400和100mhz下记录1h和
13
c nmr谱。相对于作为内标的残余溶剂峰,以百万分率报告化学位移(δ)[cdcl3:7.26(s)(对于1h),77.0(t)(对于
13
c);cd3od:4.78(s)和3.31(五重峰)(对于1h),49.15(七重峰)(对于
13
c);d2o:4.79(s)(对于1h)]。运行2d cosy和hsqc实验以支持归属。在brukerdaltonics esquire 3000esi光谱仪上使用阳性模式,以电喷雾电离模式记录低分辨率质谱(lrms)。
[0177]
合成
[0178]
萘并三唑合成
[0179][0180]
方案1.“5-乙酰氨基-2,6-脱水-3,4,5-三脱氧基-4-(3h-萘并[1,2-d][1,2,3]三唑-3-基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1778-64)”的合成.
[0181]
向叠氮化物衍生物1(60mg,0.13mmol)于无水乙腈(3ml)中的溶液中添加氟化铯(40mg,0.26mmol),之后添加1-(三甲基甲硅烷基)-2-萘基三氟甲烷磺酸盐(55μl,0.197mmol),并且将反应混合物在rt在氩气下搅拌过夜。添加饱和nahco3水溶液(20ml),并且将混合物用乙酸乙酯(100ml)萃取。将有机层分离,用水、盐水洗涤,然后经无水na2so4干燥。将干燥的有机溶剂在真空下浓缩,并且通过硅胶色谱,使用乙酸乙酯:己烷(6:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈蓬松白色粉末的纯的脱保护产物ie1778-64(38mg,64%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.62(s,3h),3.63-3.76(m,2h),3.93(dd,j=12.1,2.7hz,1h),4.07(ddd,j=9.3,6.3,
2.7hz,1h),4.58(t,j=10.3hz,1h),4.67-4.74(m,1h),5.91(dd,j=9.7,2.3hz,1h),6.12(d,j=2.3hz,1h),7.60-7.73(m,2h),7.74-7.83(m,1h),7.90(d,j=9.1hz,1h),8.03(d,j=8.0hz,1h),8.51(d,j=8.2hz,1h);
13
c nmr(101mhz,d2o):21.44,48.07,59.01,63.09,68.10,69.76,75.39,102.37,109.52,121.30,124.00,126.96,128.50,128.91,130.26,130.62,131.42,141.56,150.48,168.83,173.22;lrms[c
21h21
n4nao7](m/z):(正离子模式)487.1[m na]
[0182][0183]
方案2.关键中间体“2,6-脱水-4-叠氮基-3,4,5-三脱氧基-5-[0184]
(2,2,2-三氟乙酰氨基)-d-甘油-d-半乳-壬-2-烯酸酯(ie1530-57)”的一步合成.
[0185]
向叠氮化物衍生物1(100mg,0.22mmol)于乙腈(0.5ml)中的溶液中添加三氟乙酸酐(220μl,1.54mmol)并且将混合物在微波反应器中在135℃加热10min。在冷却后,添加meoh(1ml),并且将反应混合物在真空下浓缩。将粗制产物通过硅胶色谱,使用己烷:丙酮(3:2)作为溶剂加以纯化,得到纯的ie1530-57(84mg,75%)。1h nmr(400mhz,cdcl3):δ2.05(s,3h),2.08(s,3h),2.14(s,3h),3.82(s,3h),3.92(q,j=8.9hz,1h),4.19(dd,j=12.5,6.5hz,1h),4.47-4.57(m,2h),4.66(dd,j=12.5,2.7hz,1h),5.31(td,j=6.0,5.4,2.8hz,1h),5.39(dd,j=5.2,2.3hz,1h),6.02(d,j=2.7hz,1h),7.22(d,j=8.8hz,1h);
13
c nmr(101mhz,cdcl3):δ20.61,20.66,20.87,49.05,52.77,57.03,61.79,67.54,70.84,75.01,107.05,115.39(q,j=288.0hz),145.25,157.66(d,j=38.3hz),161.25,170.50,170.65,170.92;lrms[c
18h21
f3n4o
10
](m/z):(正离子模式)533.2[m na]
[0186][0187]
方案3.“2,6-脱水-3,4,5-三脱氧基-5-(2,2,2-三氟乙酰氨基)-4-(3h-萘并[1,2-d][1,2,3]三唑-3-基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1778-74)”的合成.
[0188]
向叠氮化物衍生物ie1530-57(60mg,0.118mmol)于无水乙腈(3ml)中的溶液中添加氟化铯(36mg,0.24mmol),之后添加1-(三甲基甲硅烷基)-2-萘基三氟甲烷磺酸盐(50μl,0.177mmol),并且将反应混合物在rt在氩气下搅拌过夜。添加饱和nahco3水溶液(20ml),并且将混合物用乙酸乙酯(100ml)萃取。将有机层分离,用水、盐水洗涤,然后经无水na2so4干燥。将干燥的有机溶剂在真空下浓缩,并且通过硅胶色谱,使用己烷:丙酮(2:1)作为溶剂加
以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加三乙胺(1ml)。将温度逐渐升至rt并且将混合物在rt搅拌过夜。将溶液在真空下浓缩,并且使用0.05m naoh将ph调整到8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈蓬松浅黄色粉末的纯的脱保护产物ie1778-74(36mg,59%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ3.63-3.75(m,2h),3.93(dd,j=12.0,2.7hz,1h),4.08(ddd,j=9.4,6.5,2.7hz,1h),4.66-4.73(m,1h),4.75(s,1h),5.99(dd,j=9.5,2.3hz,1h),6.14(d,j=2.2hz,1h),7.67-7.77(m,2h),7.81(ddd,j=8.2,7.1,1.2hz,1h),7.95(d,j=9.2hz,1h),8.07(d,j=8.0hz,1h),8.53-8.60(m,1h);
13
c nmr(101mhz,d2o):δ49.49,58.86,58.95,63.10,68.27,69.78,75.56,102.41,109.70,121.34,124.01,127.03,128.54,128.97,130.33,130.74,131.46,141.63,150.56,168.77;lrms[c
21h18
f3n4nao7](m/z):(正离子模式)541.1[m na]
[0189]
吲唑合成
[0190][0191]
方案4.“5-乙酰氨基-2,6-脱水-4-(2h-吲唑-2-基)-3,4,5-三脱氧基-d-甘油-d-半乳-壬-2-烯酸钠(ie1530-66)”的合成.
[0192]
向胺衍生物2(60mg,0.14mmol)于无水乙腈(1ml)中的溶液中添加2-叠氮基苯甲醛(30mg,0.21mmol)并且将反应混合物在135℃在微波辐照下加热15min。将混合物静置冷却到rt,在真空下浓缩,并通过硅胶色谱,使用乙酸乙酯:己烷(4:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈蓬松粉末的纯的脱保护产物ie1530-66(33mg,58%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.83(s,3h),3.63-3.73(m,2h),3.92(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.7hz,1h),4.47-4.61(m,2h),5.40-5.47(m,1h),5.93(d,j=2.2hz,1h),7.18(ddd,j=8.5,6.6,0.9hz,1h),7.41(ddd,j=8.8,6.7,1.1hz,1h),7.67(d,j=8.8hz,1h),7.79(d,j=8.5hz,1h),8.35(s,1h);
13
c nmr(101mhz,d2o):21.61,48.98,61.78,63.10,68.18,69.78,75.39,103.54,116.02,120.94,122.01,124.70,127.28,148.42,149.77,169.08,173.46;lrms[c
18h20
n3nao7](m/z):(正离子模式)436.2[m na]
[0193][0194]
方案5.中间体胺(1530-61)和最终抑制剂(ie1530-74)的合成
[0195]
7,8,9-三-o-乙酰基-4-氨基-2,6-脱水-3,4,5-三脱氧基-5-(2,2,2-三氟乙酰氨基)-d-甘油-d-半乳-壬-2-烯酸甲酯(ie1530-61)
[0196][0197]
向叠氮化物衍生物ie1530-56(200mg,0.41mmol)于乙醇(5ml)中的溶液中添加lindlar催化剂(20mg)并且将反应混合物在h2气氛下在rt搅拌过夜。在反应完成后,将反应混合物通过硅藻土床过滤,之后用乙醇(50ml)洗涤。将合并的滤液和洗涤物合并且在真空下浓缩,得到具有良好纯度的粗制ie1530-61(定量产率),其未经进一步纯化即用于下一步骤中。lrms[c
18h26
n2o
10
](m/z):(正离子模式)507.2[m na]
[0198]
2,6-脱水-3,4,5-三脱氧基-4-(2h-吲唑-2-基)-5-(2,2,2-三氟乙酰氨基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1530-74)
[0199][0200]
向胺衍生物ie1530-61(50mg,0.103mmol)于无水乙腈(1ml)中的溶液中添加2-叠氮基苯甲醛(22mg,0.15mmol)并且将反应混合物在135℃在微波辐照下加热15min。将混合物静置冷却到rt,在真空下浓缩,并通过硅胶色谱,使用乙酸乙酯:己烷(3:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加三乙胺(1ml)。将温度逐渐升至rt并且将混合物在rt搅拌过夜。将溶液在真空下浓缩,并且使用0.05m naoh将ph调整到8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,得到纯的脱保护产物ie1530-74(26mg,55%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ3.63-3.73(m,2h),3.92(dd,j=12.0,2.7hz,1h),4.05(ddd,j=9.4,6.4,2.7hz,1h),4.58-4.77(m,2h),5.54(dd,j=9.4,2.3hz,1h),5.99(d,j=2.2hz,1h),7.19(ddd,j=8.5,6.7,0.9hz,1h),7.42(ddd,j
=8.8,6.6,1.1hz,1h),7.66(dd,j=8.8,1.0hz,1h),7.79(d,j=8.5hz,1h),8.39(d,j=1.0hz,1h);
13
c nmr(101mhz,d2o):49.79,61.29,63.05,68.21,69.73,74.93,103.18,111.19-119.63(m),120.94,121.46,122.16,124.69,127.44,148.57,150.06,158.32(q,j=38.0,37.1hz),168.85;lrms[c
18h17
f3n3nao7](m/z):(正离子模式)490.2[m na]
[0201][0202]
方案6.“5-乙酰氨基-2,6-脱水-4-(2h-苯并[g]吲唑-2-基)-3,4,5-三脱氧基-d-甘油-d-半乳-壬-2-烯酸钠(ie1530-65)”的合成.
[0203]
向胺衍生物2(60mg,0.14mmol)于无水乙腈(1ml)中的溶液中添加1-叠氮基-2-萘甲醛(41mg,0.21mmol)并且将反应混合物在135℃在微波辐照下加热15min。将混合物静置冷却到rt,在真空下浓缩,并通过硅胶色谱,使用乙酸乙酯:己烷(3:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈蓬松粉末的纯的脱保护产物ie1530-65(36mg,56%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.81(s,3h),3.63-3.75(m,2h),3.93(dd,j=12.0,2.7hz,1h),4.04(ddd,j=9.3,6.3,2.7hz,1h),4.51(t,j=10.2hz,1h),4.56-4.64(m,1h),5.43(dd,j=9.5,2.3hz,1h),5.97(d,j=2.2hz,1h),7.43(d,j=9.0hz,1h),7.56-7.69(m,3h),7.86-7.95(m,1h),8.27(s,1h),8.43-8.50(m,1h);
13
c nmr(101mhz,d2o):δ21.62,49.09,61.54,63.11,68.19,69.79,75.48,103.51,118.69,118.89,121.74,123.62,124.05,125.21,126.93,127.28,128.65,132.49,145.82,149.94,169.13,173.48;lrms[c
22h22
n3nao7](m/z):(正离子模式)486.2[m na]
[0204][0205]
方案7.“2,6-脱水-4-(2h-苯并[g]吲唑-2-基)-3,4,5-三脱氧基-5-(2,2,2-三氟乙酰氨基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1530-69)”的合成.
[0206]
向胺衍生物ie1530-61(50mg,0.103mmol)于无水乙腈(1ml)中的溶液中添加1-叠氮基-2-萘甲醛(30mg,0.15mmol)并且将反应混合物在135℃在微波辐照下加热15min。将混合物静置冷却到rt,在真空下浓缩,并通过硅胶色谱,使用乙酸乙酯:己烷(2:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加三乙胺(1ml)。将温度逐渐升至rt并且将混合物在rt搅拌过夜。将溶液在真空下浓缩,并且使用0.05m naoh将ph调整到8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,得到纯的脱保护产物ie1530-69(28mg,59%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ3.64-3.74(m,2h),3.93(dd,j=11.9,2.7hz,1h),4.06(ddd,j=9.4,6.4,2.7hz,1h),4.59(t,j=10.2hz,1h),4.69-4.76(m,1h),5.52(d,j=8.8hz,1h),6.04(d,j=2.2hz,1h),7.45(d,j=9.1hz,1h),7.63(dd,j=19.3,8.1hz,3h),7.91(d,j=7.2hz,1h),8.31(s,1h),8.45(d,j=8.2hz,1h);
13
c nmr(101mhz,d2o):δ50.17,61.11,63.08,68.24,69.75,75.09,115.53(q,j=286.1,285.1hz),118.83,121.84,123.79,123.99,125.03,126.94,127.36,128.64,132.54,145.98,150.30,158.37(d,j=37.7hz),168.92;lrms[c
22h19
f3n3nao7](m/z):(正离子模式)540.2[m na]
[0207]
替代吲唑合成
[0208]
用于合成吲唑的前述方法包括在微波反应器中在高温加热胺与2-叠氮基-1-甲醛衍生物,以影响在单个步骤中的亚胺形成和环化。发现该方法不能用于所有2-叠氮基-1-甲醛衍生物。因此,开发下文所示的铜催化的一锅合成:
[0209][0210]
方案8.ie1993-8的合成
[0211]
实验程序:
[0212]
在氩气下向胺(50mg,0.103mm)、2-叠氮基-1-甲醛衍生物(1.2eq)、cui(0.1eq)和4a分子筛(50mg)于无水1,4-二烷(2ml)中的混合物中添加四甲基乙二胺(tmeda,1.0eq)。将混合物在rt搅拌1h,然后经硅藻土过滤,浓缩并通过快速二氧化硅柱色谱纯化。
[0213][0214]
方案9.吲唑的一般合成
[0215]
吲唑的一般合成(方案9)
[0216]
在氩气下向胺(1.0eq)、2-叠氮基-1-甲醛衍生物(1.2eq)、cui(0.1eq)和4a分子筛(50mg)于无水1,4-二烷(2ml)中的混合物中添加四甲基乙二胺(tmeda,1.0eq)。将混合物在rt搅拌1h,然后经硅藻土过滤,浓缩并通过快速二氧化硅柱色谱纯化,得到受保护的吲唑产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃下向该悬浮液中逐滴添加et3n(1.0ml),并且将温度逐渐升至rt且将混合物在rt搅拌过夜。将溶液在真空下浓缩,然后通过快速二氧化硅柱色谱加以纯化,得到纯的脱保护产物。
[0217]
ie1963-108
[0218][0219]1h nmr(400mhz,cd3od):δ3.56(d,j=9.4hz,1h),3.68(dd,j=11.5,5.5hz,1h),3.84(dd,j=11.5,2.9hz,1h),3.95(ddd,j=9.4,5.4,2.9hz,1h),4.63-4.72(m,2h),5.57(td,j=5.0,2.2hz,1h),5.86(d,j=2.1hz,1h),7.16(dd,j=8.6,7.2hz,1h),7.25(d,j=7.1hz,1h),7.54-7.59(m,1h),8.28(d,j=0.9hz,1h);
13
c nmr(101mhz,cd3od):δ49.78,61.34,63.39,68.69,69.92,75.03,101.18,112.68,115.73(q,j=287.2hz),116.05,123.53,123.60,124.12,126.76,148.69,151.06,157.34(q,j=37.6hz),167.81;lrms[c
18h17
brf3n3o7](m/z):(正离子模式)569.4[m na]
。
[0220]
ie1963-109
[0221][0222]1h nmr(400mhz,cd3od):δ3.56(d,j=9.4hz,1h),3.68(dd,j=11.5,5.4hz,1h),3.84(dd,j=11.5,2.9hz,1h),3.95(ddd,j=9.5,5.4,2.9hz,1h),4.64-4.73(m,2h),5.56-5.65(m,1h),5.84(d,j=2.1hz,1h),7.42(dd,j=9.0,1.5hz,1h),7.72(dt,j=9.0,1.0hz,1h),8.27(t,j=1.2hz,1h),8.54(d,j=0.9hz,1h);
13
c nmr(101mhz,cd3od):δ49.80,61.70,63.38,68.64,69.89,75.04,100.91,104.58,115.73(q,j=287.2hz),118.37,119.30,121.02,125.13,126.40,128.75,148.60,151.24,157.33(d,j=37.4hz),167.69;lrms[c
19h17
f3n4o7](m/z):(正离子模式)514.5[m na]
。
[0223]
ie1963-110
[0224][0225]1h nmr(400mhz,cd3od):δ3.56(d,j=9.5hz,1h),3.67(dd,j=11.5,5.5hz,1h),3.84(dd,j=11.5,2.9hz,1h),3.95(ddd,j=9.5,5.5,2.8hz,1h),4.65-4.72(m,2h),5.62(td,j=5.0,2.2hz,1h),5.84(d,j=2.1hz,1h),7.23(dd,j=8.7,1.3hz,1h),7.87(dd,j=8.7,1.0hz,1h),8.10(d,j=1.1hz,1h),8.45(d,j=1.0hz,1h);
13
c nmr(101mhz,cd3od):δ49.83,61.81,63.41,68.68,69.88,75.06,101.04,109.07,115.70(d,j=287.1hz),119.03,121.70,122.45,123.36,123.79,123.96,146.84,151.14,157.28(q,j=37.3hz),167.60;lrms[c
19h17
f3n4o7](m/z):(正离子模式)514.5[m na]
。
[0226]
ie1993-25
[0227][0228]1h nmr(400mhz,cd3od):δ3.54(d,j=9.5hz,1h),3.67(dd,j=11.5,5.5hz,1h),3.84(dd,j=11.5,2.9hz,1h),3.94(ddd,j=9.0,5.5,2.8hz,1h),4.64-4.70(m,2h),5.49-5.56(m,1h),5.82(d,j=2.2hz,1h),6.79(dd,j=8.9,1.9hz,1h),7.18-7.24(m,1h),7.71(dd,j=8.8,0.8hz,1h),8.27(s,1h);
13
c nmr(101mhz,cd3od):δ49.69,61.15,63.45,68.75,69.88,75.00,101.34,104.22,113.78(d,j=104.8hz),115.37,119.85,122.26,123.39,138.61,148.82,150.99,157.25(d,j=37.5hz),167.83;lrms[c
18h17
f4n3o7](m/z):(正离子模式)484.0[m na]
。
[0229]
ie1993-26
[0230][0231]1h nmr(400mhz,cd3od):δ3.56(d,j=9.3hz,1h),3.67(dd,j=11.5,5.5hz,1h),3.80-3.89(m,4h),3.9-3.97(m,4h),4.62-4.75(m,2h),5.45-5.55(m,1h),5.80(d,j=2.2hz,1h),6.24(d,j=8.0hz,1h),6.50(d,j=8.0hz,1h),8.19(s,1h);
13
c nmr(101mhz,cd3od):δ49.56,54.38,54.73,61.01,63.43,68.70,70.00,75.11,97.86,101.68,103.45,114.30,116.98,117.16,121.09,142.82,144.02,147.25,150.75,157.14;lrms[c
20h22
f3n3o9](m/z):(负离子模式)503.9[m-h]
。
[0232]
ie1993-27
[0233][0234]1h nmr(400mhz,cd3od):δ3.53(d,j=9.4hz,1h),3.67(dd,j=11.5,5.5hz,1h),3.84(dd,j=11.6,2.9hz,1h),3.94(ddd,j=9.1,5.6,2.8hz,1h),4.58-4.68(m,2h),5.40(d,j=7.1hz,1h),5.75-5.84(m,1h),5.92(s,2h),6.81(s,1h),6.88(s,1h),7.98(s,1h);
13
c nmr(101mhz,cd3od):δ49.55,60.58,63.44,68.75,69.93,75.01,92.69,94.79,100.92,101.89,114.33,117.31,122.18,145.71,146.07,149.63,150.70,157.24(d,j=37.1hz);lrms[c
19h18
f3n3o9](m/z):(负离子模式)487.9[m-h]
。
[0235][0236]
方案10.一般吲唑合成
[0237]
吲唑的一般合成(方案10)
[0238]
在氩气下向胺(1.0eq)、2-叠氮基-1-甲醛衍生物(1.2eq)、cui(0.1eq)和4a分子筛(50mg)于无水1,4-二烷(2ml)中的混合物中添加四甲基乙二胺(tmeda,1.0eq)。将混合物在rt搅拌1h,然后经硅藻土过滤,浓缩并通过快速二氧化硅柱色谱纯化,得到受保护的吲唑产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用10%甲醇/水作为溶剂加以纯化,得到纯的脱保护产物。
[0239]
ie1993-10
[0240][0241]1h nmr(400mhz,d2o):δ0.83(d,j=6.9hz,3h),0.93(d,j=6.9hz,3h),2.37(p,j=6.9hz,1h),3.63-3.71(m,2h),3.93(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.4,2.7hz,1h),4.55-4.66(m,2h),5.48-5.54(m,1h),5.95(d,j=2.2hz,1h),7.60(dd,j=8.7,1.4hz,1h),7.80(dd,j=8.8,1.0hz,1h),8.15(d,j=1.1hz,1h),8.39(d,j=1.0hz,1h);
13
c nmr(101mhz,d2o):δ18.39,18.49,35.02,48.64,62.00,63.10,68.26,69.83,75.41,103.43,117.57,120.59,121.99,122.60,124.69,135.35,147.94,149.75,169.08,175.72,180.56;lrms[c
21h23
n3na2o9](m/z):(正离子模式)529.2[m na]
。
[0242]
ie1993-20
[0243][0244]1h nmr(400mhz,d2o):δ0.84(d,j=6.9hz,3h),0.93(d,j=6.9hz,3h),2.38(p,j=6.9hz,1h),3.63-3.71(m,2h),3.92(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.4,2.7hz,1h),4.53-4.65(m,2h),5.51(dd,j=9.2,2.3hz,1h),5.95(d,j=2.1hz,1h),7.65
(dt,j=9.2,1.0hz,1h),7.85(dd,j=9.1,1.6hz,1h),8.34(dd,j=1.6,0.9hz,1h),8.50(d,j=1.0hz,1h)。
[0245]
苯基三唑衍生物合成
[0246][0247]
方案11.2-氨基苯基三唑中间体的合成和氨基的修饰.
[0248]
5-乙酰氨基-7,8,9-三-o-乙酰基-4-((2-氨基苯基)-1h-1,2,3-三唑-1-基)-2,6-脱水-3,4,5-三脱氧基-d-甘油-d-半乳-壬-2-烯酸甲酯(ie1398-24)
[0249][0250]
将9-叠氮基衍生物1(500mg,1.1mmol)和1-氨基-2-乙炔基苯(140μl,1.2mmol)溶解于meoh:h2o的4:1混合物(4ml)中。将五水合硫酸铜(ii)(50mg,0.22mmol)添加到混合物中,之后添加抗坏血酸钠(1.0ml新鲜制备的1m于h2o中的溶液)。将混合物在45℃搅拌过夜,然后静置冷却至rt。然后将混合物用dcm(200ml)稀释,用10%nh4oh(100ml)洗涤,之后用盐水(100ml)洗涤。将有机层经无水na2so4干燥并在真空下浓缩,得到粗制的受保护产物,将其通过硅胶色谱,使用乙酸乙酯:己烷(3:1)作为溶剂加以浓缩,以77%产率得到纯的ie1398-24(484mg,0.84mmol)。1h nmr(400mhz,cdcl3):δ1.70(s,3h),2.04(s,3h),2.05(s,3h),2.07(s,3h),3.80(s,3h),4.19(dd,j=12.4,7.4hz,1h),4.37(q,j=10.0hz,1h),4.67(dd,j=10.8,1.9hz,1h),4.73(dd,j=12.5,2.6hz,1h),5.35(ddd,j=7.5,4.7,2.6hz,1h),5.53(dd,j=4.6,1.8hz,1h),5.67(dd,j=10.1,2.4hz,1h),6.03(d,j=2.3hz,1h),6.65-6.76(m,2h),6.97(d,j=9.2hz,1h),7.08(ddd,j=8.3,7.2,1.6hz,1h),7.30(dd,j=7.7,1.5hz,1h),7.84(s,1h);
13
c nmr(101mhz,cdcl3):δ20.69,20.82,20.95,22.72,48.09,
52.71,58.40,62.22,67.85,71.39,76.90,107.36,113.54,116.72,117.78,119.35,128.28,129.43,144.82,145.75,148.14,161.32,170.13,170.51,170.76,171.05;lrms[c
26h31
n5o
10
](m/z):(正离子模式)596.2[m na]
[0251]
5-乙酰氨基-2,6-脱水-3,4,5-三脱氧基-4-(2-(甲基磺酰氨基)苯基)-1h-1,2,3-三唑-1-基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1778-12)
[0252][0253]
向ie1398-24(50mg,0.087mmol)于无水二氯甲烷(3ml)中的溶液中添加二异丙基乙胺(46μl,0.26mmol),之后添加甲基磺酰氯(7.0μl,0.096mmol)。将混合物在rt搅拌3h,在真空下浓缩,得到粗制产物,将其通过硅胶色谱,使用乙酸乙酯:己烷(3:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈蓬松粉末的纯的脱保护产物ie1778-12(28mg,60%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.93(s,3h),2.84(s,3h),3.64-3.76(m,2h),3.93(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.6hz,1h),4.50(dd,j=10.9,9.5hz,1h),4.57-4.64(m,1h),5.59(dd,j=9.7,2.3hz,1h),5.95(d,j=2.2hz,1h),7.12(ddd,j=7.8,5.7,2.8hz,1h),7.28-7.42(m,2h),7.95(dt,j=7.7,1.3hz,1h),8.66(s,1h);
13
c nmr(101mhz,d2o):δ21.77,39.74,48.72,59.61,63.11,68.16,69.76,75.42,102.39,121.36,123.73,123.97,123.99,127.76,129.27,143.92,145.41,150.14,168.86,173.75;lrms[c
20h24
n5nao9s](m/z):(正离子模式)556.1[m na]
[0254]
5-乙酰氨基-2,6-脱水-3,4,5-三脱氧基-4-(2-(2-羟基乙酰氨基)苯基)-1h-1,2,3-三唑-1-基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1778-25)
[0255]
[0256]
向ie1398-24(50mg,0.087mmol)于无水二氯甲烷(3ml)中的溶液中添加二异丙基乙胺(46μl,0.26mmol),之后添加乙酰氧基乙酰氯(10μl,0.096mmol)。将混合物在rt搅拌3h,在真空下浓缩,得到粗制产物,将其通过硅胶色谱,使用乙酸乙酯:己烷(4:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈蓬松粉末的纯的脱保护产物ie1778-25(33mg,74%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.93(s,3h),3.63-3.77(m,2h),3.93(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.7hz,1h),4.22(s,2h),4.47(dd,j=10.9,9.6hz,1h),4.60(dd,j=10.9,1.3hz,1h),5.61(dd,j=9.6,2.3hz,1h),5.89(d,j=2.2hz,1h),7.45(td,j=7.6,1.4hz,1h),7.53(td,j=7.8,1.7hz,1h),7.76(ddd,j=14.6,7.8,1.4hz,2h),8.33(s,1h);
13
c nmr(101mhz,d2o):δ21.68,48.73,60.06,61.33,63.08,68.07,69.72,75.39,101.85,122.10,123.92,125.54,127.13,129.18,129.63,132.87,145.31,150.53,168.75,173.65,174.10;lrms[c
21h24
n5nao9](m/z):(正离子模式)536.2[m na]
[0257][0258]
方案12.2-氨基苯基三唑衍生物处延伸的叠氮化物.
[0259]
5-乙酰氨基-7,8,9-三-o-乙酰基-2,6-脱水-4-(2-(6-叠氮基己酰氨基)苯基)-1h-1,2,3-三唑-1-基)-3,4,5-三脱氧基-d-甘油-d-半乳-壬-2-烯酸甲酯(ie1826-5)
[0260][0261]
在氩气下向2-氨基苯基三唑衍生物ie1398-24(400mg,0.70mmol)、6-叠氮基己酸(113μl,0.77mmol)和(600mg,1.40mmol)于无水dmf(10ml)中的混合物中添加diea(370μl,2.1mmol)。将混合物在rt搅拌过夜,然后在真空下浓缩,得到粗制产物,将其通过硅
胶色谱,使用乙酸乙酯:己烷(3:1)作为溶剂加以纯化,以86%产率得到纯的ie1826-5(430mg,0.60mmol)。1h nmr(400mhz,meoh-d4):δ1.41(ddt,j=9.0,6.7,3.2hz,2h),1.54-1.63(m,2h),1.67-1.77(m,5h),1.89-2.06(m,11h),2.41(t,j=7.4hz,2h),3.77(s,3h),4.11(dd,j=12.5,6.2hz,1h),4.42(t,j=10.2hz,1h),4.57(ddd,j=15.2,11.6,2.4hz,2h),5.34(td,j=6.3,2.7hz,1h),5.47-5.57(m,2h),6.11(d,j=2.5hz,1h),7.12(td,j=7.6,1.3hz,1h),7.27(ddd,j=8.6,7.4,1.6hz,1h),7.62(dd,j=8.0,1.6hz,1h),8.17(d,j=8.0hz,1h),8.37(s,1h);
13
c nmr(101mhz,meoh-d4):δ19.25,19.32,19.36,21.14,24.84,25.98,28.24,37.19,37.24,50.88,51.71,59.66,61.77,67.32,70.21,76.50,106.75,119.86,121.01,122.38,124.24,127.79,128.45,135.38,146.10,146.75,170.01,170.10,171.00,171.80,172.95;lrms[c
32h40
n8o
11
](m/z):(正离子模式)735.5[m na]
[0262]
5-乙酰氨基-2,6-脱水-4-(2-(6-叠氮基己酰氨基)苯基)-1h-1,2,3-三唑-1-基)-3,4,5-三脱氧基-d-甘油-d-半乳-壬-2-烯酸钠(ie1826-23)
[0263][0264]
将ie1826-5(40mg,0.056mmol)悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈白色粉末的纯的脱保护产物ie1826-23(23mg,71%产率)。1h nmr(400mhz,d2o):δ1.35-1.46(m,2h),1.59-1.73(m,4h),1.94(s,3h),2.44(t,j=7.4hz,2h),3.34(t,j=6.8hz,2h),3.64-3.73(m,2h),3.93(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.4,6.4,2.7hz,1h),4.45(t,j=10.2hz,1h),4.60(d,j=11.0hz,1h),5.61(dd,j=9.6,2.3hz,1h),5.88(d,j=2.2hz,1h),7.43-7.57(m,3h),7.69-7.76(m,1h),8.24(s,1h);
13
c nmr(101mhz,d2o):δ21.73,24.63,25.47,27.64,35.90,48.78,50.94,60.04,63.09,68.07,69.70,75.38,101.61,121.99,125.35,126.96,127.53,129.41,129.72,133.37,145.15,150.71,168.58,173.64,176.15;lrms[c
25h31
n8nao8](m/z):(正离子模式)617.3[m na]
[0265][0266]
方案13.2-氨基苯基三唑衍生物处延伸的三唑.
[0267]
一般程序:
[0268]
将叠氮化物中间体ie1826-5(50mg,0.07mmol)和适当的炔(0.084mmol)溶解于meoh:h2o的4:1混合物(4ml)中。将五水合硫酸铜(ii)(3mg,0.014mmol)添加到混合物中,之后添加抗坏血酸钠(0.1ml新鲜制备的1m于h2o中的溶液)。将混合物在60℃搅拌6h,然后静置冷却至rt。然后将混合物用dcm(200ml)稀释,用10%nh4oh(100ml)洗涤,之后用盐水(100ml)洗涤。将有机层经无水na2so4干燥并在真空下浓缩,得到粗制的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,得到纯的脱保护三唑。
[0269]
5-乙酰氨基-2,6-脱水-3,4,5-三脱氧基-4-(2-(6-(4-苯基-1h-1,2,3-三唑-1-基)己酰氨基)苯基)-1h-1,2,3-三唑-1-基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1826-30)
[0270][0271]1h nmr(400mhz,d2o):δ1.25(dt,j=14.3,7.0hz,2h),1.67(p,j=7.1hz,2h),1.88(s,3h),1.91-1.98(m,2h),2.37(t,j=7.0hz,2h),3.63-3.73(m,2h),3.91(dd,j=12.0,2.6hz,1h),4.02(ddd,j=9.3,6.2,2.7hz,1h),4.35-4.46(m,3h),4.54(dd,j=11.2,1.3hz,1h),5.47(dd,j=9.7,2.3hz,1h),5.79(d,j=2.2hz,1h),7.27-7.33(m,2h),7.39-7.53(m,5h),7.59(dd,j=7.8,1.9hz,2h),7.97(s,1h),8.15(s,1h);
13
c nmr(101mhz,d2o):
δ21.71,24.14,24.49,28.62,35.84,48.72,50.15,59.92,63.08,68.05,69.69,75.35,101.51,121.68,121.90,123.43,125.41,125.56,126.81,128.65,129.05,129.11,129.31,129.40,133.29,145.29,147.23,150.73,168.55,173.51,175.54;lrms[c
33h37
n8nao8](m/z):(正离子模式)719.4[m na]
[0272]
5-乙酰氨基-2,6-脱水-4-(2-(6-(4-羧基-1h-1,2,3-三唑-1-基)己酰氨基)苯基)-1h-1,2,3-三唑-1-基)-3,4,5-三脱氧基-d-甘油-d-半乳-壬-2-烯酸二钠(ie1826-34)
[0273][0274]1h nmr(400mhz,d2o):δ1.32(qd,j=8.6,6.0hz,2h),1.64-1.74(m,2h),1.91(s,3h),1.97(q,j=7.3hz,2h),2.40(t,j=7.4hz,2h),3.65-3.76(m,2h),3.92(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.6hz,1h),4.42-4.52(m,3h),4.60(dd,j=11.0,1.3hz,1h),5.60(dd,j=9.7,2.3hz,1h),5.87(d,j=2.3hz,1h),7.42-7.55(m,3h),7.71(dd,j=7.7,1.7hz,1h),8.22(d,j=3.8hz,2h);
13
c nmr(101mhz,d2o):δ21.71,24.46,24.98,28.91,35.72,48.78,50.24,60.01,63.08,68.06,69.72,75.39,101.69,122.02,125.37,126.94,127.08,127.56,129.37,129.73,133.28,144.67,145.14,150.65,167.67,168.63,173.59,176.00;lrms[c
28h32
n8na2o
10
](m/z):(正离子模式)709.3[m na]
。
[0275]
5-乙酰氨基-2,6-脱水-3,4,5-三脱氧基-4-(2-(6-(4-(甲氧基甲基)-1h-1,2,3-三唑-1-基)己酰氨基)苯基)-1h-1,2,3-三唑-1-基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1826-38)
[0276][0277]1h nmr(400mhz,d2o):δ1.27(td,j=8.8,4.5hz,2h),1.67(p,j=7.3hz,2h),1.93(d,j=9.7hz,5h),2.40(t,j=7.1hz,2h),3.33(s,3h),3.68(dd,j=11.9,6.3hz,1h),3.73(d,j=9.7hz,1h),3.92(dd,j=11.8,2.4hz,1h),4.03(ddd,j=9.3,6.1,2.4hz,1h),4.41-4.49(m,3h),4.53(s,2h),4.60(d,j=10.9hz,1h),5.60(dd,j=9.8,2.2hz,1h),5.86(d,j=1.8hz,1h),7.42-7.54(m,3h),7.72(d,j=7.5hz,1h),8.00(s,1h),8.22(s,1h);
13
c nmr
(101mhz,d2o):δ21.71,24.35,24.84,28.83,35.75,48.79,50.13,57.26,60.03,63.08,64.28,68.06,69.69,75.38,101.60,121.95,124.94,124.99,126.70,127.43,129.30,129.66,133.31,143.58,145.14,150.72,168.56,173.59,175.86;lrms[c
29h37
n8nao9](m/z):(正离子模式)687.3[m na]
。
[0278]
5-乙酰氨基-2,6-脱水-3,4,5-三脱氧基-4-(2-(6-(4-(羟基甲基)-1h-1,2,3-三唑-1-基)己酰氨基)苯基)-1h-1,2,3-三唑-1-基)-d-甘油-d-半乳-壬-2-烯酸钠(ie1826-44)
[0279][0280]1h nmr(400mhz,d2o):δ1.22-1.34(m,2h),1.67(p,j=7.5hz,2h),1.88-2.00(m,5h),2.40(t,j=7.3hz,2h),3.65-3.76(m,2h),3.93(dd,j=11.9,2.7hz,1h),4.03(ddd,j=9.4,6.5,2.6hz,1h),4.40-4.50(m,3h),4.60(d,j=10.9hz,1h),4.66(s,2h),5.60(dd,j=9.7,2.3hz,1h),5.86(d,j=2.2hz,1h),7.43-7.55(m,3h),7.68-7.76(m,1h),7.96(s,1h),8.22(s,1h);
13
c nmr(101mhz,d2o):δ21.72,24.38,24.89,28.87,35.75,48.78,50.09,54.55,60.02,63.08,68.06,69.71,75.39,101.63,122.01,123.97,125.12,126.79,127.47,129.32,129.69,133.31,145.13,146.65,150.69,168.57,173.61,175.91;lrms[c
28h35
n8nao9](m/z):(正离子模式)673.1[m na]
。
[0281]
二聚体合成和表征
[0282][0283]
方案14.二聚体ie1826-1的合成.
[0284]
ie1826-1
[0285][0286]
在氩气下向ie1398-24(50mg,0.087mmol)于无水二氯甲烷(3ml)中的溶液中添加二异丙基乙胺(46μl,0.26mmol),之后添加十二烷二酰基二氯化物(11μl,0.043mmol)。将混合物在rt搅拌3h,在真空下浓缩,得到粗制产物,将其通过硅胶色谱,使用乙酸乙酯:己烷(4:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈白色粉末的纯的脱保护产物ie1826-1(32mg,66%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.26(q,j=7.8,5.9hz,12h),1.60(q,j=6.8hz,4h),1.91(s,6h),2.33-2.43(m,4h),3.64-3.75(m,4h),3.92(dd,j=12.0,2.7hz,2h),4.02(ddd,j=9.3,6.2,2.7hz,2h),4.43(dd,j=11.2,9.3hz,2h),4.58(dd,j=10.8,1.4hz,2h),5.57(dd,j=9.6,2.4hz,2h),5.85(d,j=2.1hz,2h),7.39(td,j=7.5,1.4hz,2h),7.47(td,j=7.7,1.6hz,2h),7.55-7.62(m,2h),7.65(dd,j=7.7,1.6hz,2h),8.20(s,2h);
13
c nmr(101mhz,d2o):δ21.74,24.99,27.99,28.15,36.24,48.78,59.96,63.09,68.08,69.69,75.36,101.56,121.99,124.56,126.37,127.18,129.27,129.62,133.53,145.27,150.70,168.52,173.59,176.39;lrms[c
50h62n10
na2o
16
](m/z):(正离子模式)1128.0.0[m na]
[0287][0288]
方案15.二聚体ie1826-14的合成
[0289]
ie1826-14
[0290][0291]
将叠氮化物衍生物ie1826-5(40mg,0.056mmol)和庚二炔(3.0μl,0.028mmol)溶解于meoh:h2o的4:1混合物(4ml)中。将五水合硫酸铜(ii)(3.0mg,0.22mmol)添加到混合物中,之后添加抗坏血酸钠(0.1ml新鲜制备的1m于h2o中的溶液)。将混合物在60℃搅拌6h,然后静置冷却至rt。然后将混合物在真空下浓缩,得到粗制产物,将其通过硅胶色谱,使用乙酸乙酯:甲醇(7:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈白色粉末的纯的脱保护产物ie1826-14(24mg,69%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.19(ddt,j=13.6,9.8,6.2hz,4h),1.62(p,j=7.4hz,4h),1.71-1.82(m,2h),1.85-1.92(m,10h),2.34(q,j=6.1,5.0hz,4h),2.51(td,j=7.7,3.0hz,4h),2.85(s,2h),3.60-3.75(m,6h),3.91(dd,j=12.0,2.7hz,2h),4.02(ddd,j=9.3,6.2,2.6hz,2h),4.34(t,j=6.7hz,4h),4.38-4.46(m,2h),4.57(dd,j=11.0,1.3hz,2h),5.55(dd,j=9.8,2.2hz,2h),5.83(d,j=2.2hz,2h),7.29-7.37(m,4h),7.40-7.48(m,2h),7.55-7.64(m,4h),8.14(d,j=9.5hz,2h);
13
c nmr(101mhz,d2o):δ21.73,23.77,24.29,24.79,27.92,28.80,35.94,48.77,49.92,59.95,63.09,68.07,69.70,75.38,101.58,121.83,123.01,123.82,125.86,126.91,128.91,129.41,133.39,145.23,147.53,150.73,168.52,173.51,175.47;lrms[c
57h70n16
na2o
16
](m/z):(正离子模式)1304.3[m na]
[0292][0293]
方案16.二聚体ie1826-28的合成
[0294]
5-乙酰氨基-7,8,9-三-o-乙酰基-2,6-脱水-3,4,5-三脱氧基-4-(2-(庚-6-炔酰氨基)苯基)-1h-1,2,3-三唑-1-基)-d-甘油-d-半乳-壬-2-烯酸甲酯(ie1826-20)
[0295][0296]
在氩气下向2-氨基苯基三唑衍生物ie1398-24(200mg,0.35mmol)、7-庚炔酸(55μl,0.42mmol)和(300mg,0.7mmol)于无水dmf(5ml)中的混合物中添加diea(240μl,1.4mmol)。将混合物在rt搅拌过夜,然后在真空下浓缩,得到粗制产物,将其通过硅胶色谱,使用乙酸乙酯:己烷(2:1)作为溶剂加以纯化,以80%产率得到纯的ie1826-20(190mg,0.28mmol)。lrms[c
33h39
n5o
11
](m/z):(正离子模式)735.5[m na]
[0297]
ie1826-28
[0298]
[0299]
将叠氮化物衍生物ie1826-5(32mg,0.044mmol)和炔衍生物ie1826-20(30mg,0.044mmol)溶解于meoh:h2o的4:1混合物(4ml)中。将五水合硫酸铜(ii)(2.5mg,0.01mmol)添加到混合物中,之后添加抗坏血酸钠(0.1ml新鲜制备的1m于h2o中的溶液)。将混合物在60℃搅拌6h,然后静置冷却至rt。然后将混合物在真空下浓缩,得到粗制产物,将其通过硅胶色谱,使用乙酸乙酯:甲醇(10:1)作为溶剂加以纯化,得到纯的受保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用2%乙腈/水作为溶剂加以纯化,在冷冻干燥后得到呈白色粉末的纯的脱保护产物ie1826-28(17mg,65%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.18-1.25(m,2h),1.60(q,j=7.7,7.2hz,6h),1.85-1.96(m,8h),2.34(dt,j=14.6,7.6hz,4h),2.64-2.74(m,2h),3.60-3.72(m,4h),3.92(dd,j=12.0,2.7hz,2h),4.02(ddd,j=9.3,6.2,2.6hz,2h),4.35-4.48(m,4h),4.53-4.64(m,2h),5.49-5.59(m,2h),5.82(dd,j=12.3,2.3hz,2h),7.33-7.55(m,6h),7.57-7.63(m,1h),7.65(dd,j=7.7,1.4hz,1h),7.80(s,1h),8.10(s,1h),8.15(s,1h);lrms[c
51h61n13
na2o
16
](m/z):(正离子模式)1181.0[m na]
[0300][0301]
方案17.一般苯基三唑合成
[0302]
ie1963-85
[0303][0304]
向胺ie1398-24(60mg,0.105mmol)于吡啶(2ml)中的溶液中添加ac2o(50μl,0.52mmol),并且将反应混合物在rt在氩气下搅拌过夜。将反应混合物在真空下浓缩,并且将粗制产物通过硅胶色谱纯化,得到纯的脱保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用10%甲醇/水作为溶剂加以纯化,在冷冻干燥后得到纯的脱保护产物ie1963-85(74%产率,历经两个步骤)。1h nmr(400mhz,d2o):δ1.93(s,4h),2.15(s,3h),3.64-3.76(m,2h),3.92(dd,j=11.9,2.7hz,1h),4.03(ddd,j=9.3,6.2,2.6hz,1h),4.46(t,j=10.3hz,1h),4.60(d,j=10.9hz,1h),5.60(dd,j=9.6,2.3hz,1h),5.88(d,j=2.2hz,1h),7.44-7.54(m,3h),7.73(dd,j=7.3,1.5hz,1h),8.25(s,1h);
13
c nmr(101mhz,d2o):δ21.70,22.32,48.79,60.01,63.07,68.06,69.72,75.37,101.81,122.07,125.44,127.09,127.61,129.26,129.69,133.28,145.00,150.57,168.70,173.57,173.63;lrms[c
21h24
n5nao8](m/z):(正离子模式)519.3[m na]
。
[0305]
酰胺ie1963-41和ie1963-45的合成
[0306]
向胺ie1398-24(60mg,0.105mmol)于无水dcm(2ml)中的溶液中添加diea(90μl,0.52mmol),之后逐份添加酰氯(2eq),并且将反应混合物在rt在氩气下搅拌过夜。将反应混合物在真空下浓缩,并且将粗制产物通过硅胶色谱纯化,得到纯的脱保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用10%甲醇/水作为溶剂加以纯化,得到纯的脱保护产物。
[0307]
ie1963-41
[0308][0309]1h nmr(400mhz,d2o):δ1.79(s,3h),3.65(dq,j=11.4,5.9hz,2h),3.85-3.92(m,1h),3.98(ddd,j=9.4,6.1,2.7hz,1h),4.36(t,j=10.1hz,1h),4.54(d,j=10.9hz,1h),5.54(dd,j=9.3,2.6hz,1h),5.77(d,j=2.2hz,1h),7.34(t,j=7.7hz,1h),7.43(t,j=7.6hz,1h),7.51(d,j=7.1hz,1h),7.58(t,j=7.8hz,1h),7.64(d,j=7.7hz,1h),7.72(s,1h),7.82(dd,j=17.3,7.8hz,2h),8.27(s,1h);
13
c nmr(101mhz,d2o):δ21.53,48.69,59.82,63.06,68.03,69.66,75.33,99.99,101.39,118.92,119.92,121.45,122.91,124.83,125.63,126.73,128.69,129.44,130.73,133.70,135.42,145.94,149.00,150.69,165.53,168.48,173.47;lrms[c
27h25
f3n5nao9](m/z):(正离子模式)665.6[m na]
。
[0310]
ie1963-45
[0311][0312]1h nmr(400mhz,d2o):δ1.87(s,3h),3.64-3.73(m,2h),3.92(dd,j=12.0,2.7hz,1h),4.02(ddd,j=9.4,6.3,2.7hz,1h),4.08(s,3h),4.40(t,j=10.3hz,1h),4.53-4.61(m,1h),5.59(dd,j=9.9,2.3hz,1h),5.81(d,j=2.2hz,1h),7.35-7.42(m,1h),7.46(dd,j=7.6,1.5hz,1h),7.49-7.54(m,1h),7.72(td,j=8.1,1.5hz,2h),8.29(s,1h);
13
c nmr(101mhz,d2o):δ21.60,48.70,59.93,62.46,63.08,68.05,69.68,75.39,101.51,110.56(d,j=21.4hz),121.79,124.55,126.13,127.56,129.18,129.68,133.21,145.26,148.43,150.64,151.55,163.74,168.53,173.60,174.00;lrms[c
27h25
f3n5nao9](m/z):(正离子模式)665.6[m na]
。
[0313]
磺胺类ie1963-50和ie1963-54的合成
[0314]
向胺ie1398-24(60mg,0.105mmol)于无水吡啶(2ml)中的溶液中添加磺酰氯(1.2eq)并且将反应混合物在rt在氩气下搅拌过夜。将反应混合物在真空下浓缩,并且将粗制产物通过硅胶色谱纯化,得到纯的脱保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化
合物在c18-gracepuretm滤筒上,使用10%甲醇/水作为溶剂加以纯化,得到纯的脱保护产物。
[0315]
ie1963-50
[0316][0317]1h nmr(400mhz,d2o):δ1.93(s,3h),2.86(s,3h),3.64-3.74(m,2h),3.93(dd,j=11.9,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.6hz,1h),4.50(t,j=10.2hz,1h),4.60(d,j=10.9hz,1h),5.59(dd,j=9.7,2.4hz,1h),5.94(d,j=2.2hz,1h),7.16(ddd,j=8.3,5.0,3.6hz,1h),7.34-7.40(m,2h),7.93(dd,j=7.3,0.8hz,1h),8.65(s,1h);
13
c nmr(101mhz,d2o):δ21.77,39.67,48.72,59.66,63.11,68.15,69.76,75.42,102.34,121.93,123.89,123.92,124.06,127.91,129.33,142.91,145.41,150.17,168.85,173.75;lrms[c
20h24
n5nao9s](m/z):(正离子模式)555.5[m na]
。
[0318]
ie1963-54
[0319][0320]1h nmr(400mhz,d2o):δ1.89(s,3h),3.64-3.73(m,2h),3.93(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.7hz,1h),4.36(t,j=10.3hz,1h),4.53-4.61(m,1h),5.50(dd,j=9.9,2.3hz,1h),5.87(d,j=2.2hz,1h),7.15(td,j=7.4,1.4hz,1h),7.23(d,j=7.8hz,1h),7.28-7.34(m,1h),7.66(d,j=8.8hz,2h),7.76(dd,j=7.8,1.7hz,1h),8.17(d,j=8.8hz,2h),8.32(s,1h);
13
c nmr(101mhz,d2o):δ21.70,48.64,59.55,63.11,68.12,69.74,75.40,101.92,122.69,123.42,123.99,125.04,125.88,126.43,127.97,129.28,142.67,145.17,148.39,149.76,150.41,168.75,173.65;lrms[c
25h25
n6nao
11
s](m/z):(正离子模式)662.6[m na]
。
[0321][0322]
方案18.苯基三唑合成
[0323]
ie1826-108
[0324][0325]
向胺ie1398-24(1.0g,1.744mmol)于无水dmf(6ml)中的溶液中添加boc-gly-oh(0.61g,3.48mmol,2.0eq),之后添加diea(1.21ml,6.976mmol,4.0eq)和comu(1.49g,3.48mmol,2.0eq),并且将反应混合物在rt搅拌过夜。将混合物在真空下浓缩,并且将粗制产物通过硅胶色谱(己烷/丙酮3:2)纯化,得到纯的n-boc保护的产物。在0℃向boc保护的产物(1.0g,1.369mmol)于无水dcm中的溶液中添加tfa(2.1ml,27.37mmol,20eq)并且将反应混合物升温至rt并在氩气下搅拌过夜。将反应物用乙腈稀释,然后在冷却到0℃后,通过添加粉末碳酸钠加以淬灭并搅拌5min且过滤,用水洗涤并将有机溶剂经无水na2so4干燥并且在减压下浓缩。将粗制胺通过快速色谱(乙酸乙酯/甲醇/水;7/1/0.5)纯化,历经两个步骤以48%产率得到纯胺ie1826-108。1h nmr(400mhz,cd3od):δ1.80(s,3h),2.04(s,3h),2.05(s,3h),2.07(s,3h),3.83(s,3h),3.93(s,2h),4.18(dd,j=12.5,6.2hz,1h),4.53(t,j=10.1hz,1h),4.63(ddd,j=12.5,9.7,2.3hz,2h),5.41(td,j=6.4,2.7hz,1h),5.56(td,j=8.2,7.4,2.3hz,2h),6.18(d,j=2.2hz,1h),7.22-7.32(m,1h),7.34-7.44(m,1h),7.67(dd,j=7.8,1.6hz,1h),8.07(d,j=8.1hz,1h),8.42(s,1h);
13
c nmr(101mhz,cd3od):δ19.24,19.33,19.34,21.14,41.03,51.71,59.82,61.75,67.32,70.12,76.45,106.81,121.61,121.71,123.35,125.31,128.43,128.71,134.45,145.98,146.12,161.53,164.50,170.01,170.09,171.02,172.04;lrms[c
28h34
n6o
11
](m/z):(正离子模式)630.8[m h]
。
[0326]
酰胺ie1993-4、ie1963-114和ie1963-62的合成
[0327]
向胺ie1826-108(50mg,0.08mmol)于无水dcm(2ml)中的溶液中添加diea(90μl,0.52mmol),之后逐份添加酰氯(2eq),并且将反应混合物在rt在氩气下搅拌过夜。将反应混
合物在真空下浓缩,并且将粗制产物通过硅胶色谱纯化,得到纯的脱保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用10%甲醇/水作为溶剂加以纯化,得到纯的脱保护产物。
[0328]
ie1993-4
[0329][0330]1h nmr(400mhz,d2o):δ1.86(s,3h),3.64-3.74(m,2h),3.92(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.3,6.2,2.6hz,1h),4.16-4.29(m,2h),4.38-4.48(m,1h),4.55(d,j=11.0hz,1h),5.41(dd,j=9.8,2.4hz,1h),5.75(d,j=2.2hz,1h),7.37(t,j=7.5hz,1h),7.47(td,j=7.7,1.6hz,1h),7.56(t,j=7.6hz,2h),7.62-7.67(m,2h),7.83(d,j=8.2hz,1h),7.87(dd,j=7.4,1.8hz,2h),8.29(s,1h);
13
c nmr(101mhz,d2o):δ21.68,43.99,48.66,59.89,63.08,68.09,69.70,75.33,101.90,122.41,123.00,124.91,126.78,127.43,128.80,128.88,129.46,132.39,132.65,133.18,145.23,150.35,168.68,170.94,171.11,173.59;lrms[c
28h29
n6nao9](m/z):(正离子模式)638.4[m na]
。
[0331]
ie1963-114
[0332][0333]1h nmr(400mhz,d2o):δ1.84(s,3h),2.99(s,6h),3.68(ddd,j=14.3,9.3,3.8hz,2h),3.91(dd,j=12.0,2.7hz,1h),4.01(ddd,j=9.3,6.3,2.7hz,1h),4.15(q,j=16.8hz,2h),4.36(t,j=10.3hz,1h),4.51(dd,j=11.1,1.3hz,1h),5.05(dd,j=9.8,2.3hz,1h),5.56(d,j=2.2hz,1h),6.87(d,j=9.0hz,2h),7.38(td,j=7.6,1.4hz,1h),7.49(td,j=7.7,1.7hz,1h),7.62-7.67(m,1h),7.77(d,j=8.9hz,2h),7.85(d,j=8.1hz,1h),8.13(s,1h);
13
c nmr(101mhz,d2o):δ21.62,39.60,44.21,48.72,59.88,63.13,68.06,69.71,75.43,101.99,112.09,118.79,122.54,123.02,124.76,126.72,129.12,129.18,129.56,133.08,144.69,150.13,153.81,168.39,170.51,171.37,173.56;lrms[c
30h34
n7nao9](m/
z):(正离子模式)681.5[m na]
。
[0334]
ie1963-62
[0335][0336]1h nmr(400mhz,d2o):δ1.80(s,3h),3.29(d,j=1.7hz,2h),3.84(t,j=11.9hz,1h),3.95(s,1h),4.13-4.26(m,2h),4.35(t,j=10.1hz,1h),4.44(d,j=11.0hz,1h),5.22(d,j=9.4hz,1h),5.62(s,1h),7.36(d,j=7.8hz,1h),7.42-7.56(m,4h),7.61(d,j=7.7hz,1h),7.71(d,j=7.4hz,2h),7.78(d,j=7.5hz,2h),7.89(dd,j=14.9,8.5hz,3h),8.24(s,1h);lrms[c
34h33
n6nao9](m/z):(正离子模式)714.6[m na]
。
[0337]
磺胺类ie1993-9和ie1963-99的合成
[0338]
向胺ie1826-108(50mg,0.08mmol)于无水dcm(2ml)中的溶液中添加dmap(催化量),之后添加磺酰氯(1.2eq),并且将反应混合物在rt在氩气下搅拌过夜。将反应混合物在真空下浓缩,并且将粗制产物通过硅胶色谱纯化,得到纯的脱保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用10%甲醇/水作为溶剂加以纯化,得到纯的脱保护产物。
[0339]
ie1993-9
[0340][0341]1h nmr(400mhz,d2o):δ1.92(s,3h),3.64-3.76(m,2h),3.86(s,2h),3.93(dd,j=11.9,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.8hz,1h),4.50(t,j=10.2hz,1h),4.60(d,j=10.9hz,1h),5.63(dd,j=9.7,2.2hz,1h),5.91(d,j=2.2hz,1h),7.34-7.46(m,2h),7.61-7.76(m,3h),8.16(d,j=7.8hz,1h),8.34(dt,j=8.2,1.6hz,1h),8.37(s,1h),8.57(t,j=2.0hz,1h);
13
c nmr(101mhz,d2o):δ21.75,46.94,48.67,59.95,63.10,68.12,69.71,75.44,101.88,121.67,122.17,122.81,124.53,126.77,127.05,128.82,129.43,130.84,132.58,133.01,141.90,145.19,147.80,150.50,168.71,171.36,173.71;lrms
[c
27h28
n7nao
12
s](m/z):(正离子模式)719.5[m na]
。
[0342]
ie1963-99
[0343][0344]1h nmr(400mhz,d2o):δ1.93(s,3h),3.65-3.75(m,2h),3.85(s,2h),3.93(dd,j=11.9,2.7hz,1h),4.03(ddd,j=9.3,6.3,2.6hz,1h),4.52(dd,j=10.9,9.5hz,1h),4.61(dd,j=11.0,1.0hz,1h),5.64(dd,j=9.6,2.3hz,1h),5.92(d,j=2.2hz,1h),7.37(td,j=7.6,1.4hz,1h),7.44(td,j=7.8,1.7hz,1h),7.64(dd,j=8.1,1.3hz,1h),7.70(dd,j=7.7,1.6hz,1h),7.99(d,j=8.8hz,2h),8.28(d,j=8.8hz,2h),8.39(s,1h);
13
c nmr(101mhz,d2o):δ21.75,46.82,48.65,60.03,63.09,68.12,69.71,75.46,101.88,122.17,123.10,124.52,124.75,126.90,127.82,128.85,129.46,132.97,145.17,145.79,149.50,150.53,168.71,171.26,173.68;lrms[c
27h28
n7nao
12
s](m/z):(正离子模式)719.5[m na]
。
[0345][0346]
方案19.苯基三唑合成
[0347]
ie1826-106
[0348][0349]
将boc-β-丙氨酸-oh(333mg,1.76mmol)用于dmf(3ml)中的diea(0.61ml,3.5mmol)和comu(750mg,1.76mmol)活化,然后添加到胺ie1398-24(504mg,0.879mmol)于dmf(3ml)中
的搅拌溶液中。将混合物在rt搅拌过夜,然后在真空下浓缩。然后将粗制产物溶解于水中,用乙酸乙酯(50ml
×
4)萃取,将有机层合并,用硫酸镁干燥,在真空下浓缩,然后通过硅胶色谱,使用乙酸乙酯/己烷(4:1)加以纯化。在0℃向boc保护的产物于无水dcm(15ml)中的溶液中添加tfa(2.0ml,26.0mmol,20eq)并且将反应混合物升温至rt并在氩气下搅拌过夜。将反应物用乙腈稀释,然后在冷却到0℃后,通过添加粉末碳酸钠加以淬灭并搅拌5min且过滤,用水洗涤并将有机溶剂经无水na2so4干燥并且在减压下浓缩。将粗制胺通过快速色谱(丙酮/甲醇(4:1))纯化,历经两个步骤,以44%产率得到纯胺ie1826-106。1h nmr(400mhz,cd3od):δ1.80(s,3h),2.04(s,3h),2.05(s,3h),2.07(s,3h),2.64(t,j=6.9hz,2h),3.05(t,j=7.3hz,2h),3.83(s,3h),4.18(dd,j=12.5,6.3hz,1h),4.52(t,j=10.1hz,1h),4.61(dd,j=12.5,2.7hz,1h),4.67(dd,j=10.8,2.0hz,1h),5.41(td,j=6.4,2.7hz,1h),5.53-5.66(m,2h),6.18(d,j=2.2hz,1h),7.21(t,j=7.6hz,1h),7.30-7.40(m,1h),7.63-7.73(m,1h),8.21(d,j=8.1hz,1h),8.50(s,1h);
13
c nmr(101mhz,cd3od):δ19.25,19.33,19.38,21.18,33.72,38.98,51.72,59.70,61.78,67.33,70.13,76.46,106.88,120.02,121.68,122.56,124.47,127.89,128.48,135.17,145.98,146.59,161.56,170.00,170.10,171.01,171.39,171.83;lrms[c
29h36
n6o
11
](m/z):(正离子模式)645.3[m h]
。
[0350]
酰胺ie1963-84的合成
[0351][0352]
向胺ie1826-106(60mg,0.093mmol)于无水dcm(2ml)中的溶液中添加diea(80μl,0.47mmol),之后逐滴添加环丙烷羰酰氯(2eq),并且将反应混合物在rt在氩气下搅拌过夜。将反应混合物在真空下浓缩,并且将粗制产物通过硅胶色谱纯化,得到纯的脱保护产物。将受保护产物悬浮于meoh:h2o的1:1混合物(2ml)中。在0℃向该悬浮液中逐滴添加naoh溶液(1.0m)直到ph~14。将温度逐渐升至rt并且将混合物在rt搅拌过夜。然后将溶液用ir-120(h
)树脂酸化(到ph=5),过滤并用meoh(10ml)和h2o(10ml)洗涤。然后将合并的滤液和洗涤物在真空下浓缩,然后用蒸馏水(5ml)稀释并使用0.05m naoh调整到ph=8.0,以将化合物转化为其钠盐。最后,将化合物在c18-gracepuretm滤筒上,使用10%甲醇/水作为溶剂加以纯化,得到纯的脱保护产物。1h nmr(400mhz,d2o):δ0.76-0.85(m,4h),1.59(dq,j=7.2,6.0,5.5hz,1h),1.93(s,3h),2.57-2.67(m,2h),3.53(t,j=6.5hz,2h),3.65-3.75(m,2h),3.92(dd,j=12.0,2.7hz,1h),4.03(ddd,j=9.2,6.2,2.6hz,1h),4.45(dd,j=10.9,9.6hz,1h),4.55-4.63(m,1h),5.60(dd,j=9.6,2.3hz,1h),5.88(d,j=2.2hz,1h),7.45(td,j=7.6,1.5hz,1h),7.51(td,j=7.6,1.7hz,1h),7.59(dd,j=8.0,1.4hz,1h),7.71(dd,j=7.6,1.6hz,1h),8.31(s,1h);
13
c nmr(101mhz,d2o):δ6.64,14.03,21.72,35.83,36.24,48.78,60.03,63.07,68.06,69.70,75.36,101.74,122.13,124.73,126.52,127.39,129.32,129.65,133.25,145.33,150.62,168.66,173.31,173.60,177.22;
4.52(m,3h),4.57(dd,j=10.9,1.2hz,1h),5.56(dd,j=9.7,2.3hz,1h),5.86(d,j=2.2hz,1h),7.40-7.48(m,6h),7.51(td,j=7.7,1.7hz,1h),7.60(dd,j=7.9,1.4hz,1h),7.72(dd,j=7.7,1.7hz,1h),8.24(s,1h);
13
c nmr(101mhz,d2o):δ21.71,37.06,39.35,48.72,57.50,59.99,63.09,68.09,69.71,75.38,101.86,122.36,124.78,126.65,127.42,128.74,128.88,129.30,129.62,130.70,133.16,144.96,150.52,168.69,172.87,173.60。
[0361]
生物学
[0362]
细胞和病毒:
[0363]
在补充有1%谷氨酰胺(200mm)和2%胎牛血清的伊格尔最低必需培养基(eagle's minimal essential medium,emem)中培养llc-mk2细胞(恒河猴肾,atcc ccl-7)和ma104细胞(恒河猴肾,atcc crl-2378.1)。在hpiv-3(llc-mk2)和hpiv-1(ma104)感染和感染后孵育期间,在仅补充有1%谷氨酰胺的emem中维持llc-mk2和ma104细胞。在5%co2的加湿气氛中在37℃孵育所有细胞系。从美国典型培养物保藏中心(american type culture collection,atcc)获得hpiv-3(毒株c-243)和hpiv-1(毒株c-35)。从viratree获得hpiv-3(毒株js)。hpiv-3(毒株ci002)是从黄金海岸大学医院(gold coast university hospital)获得的临床分离物。在5%co2的加湿气氛中在35℃,将病毒在用于hpiv-3的llc-mk2细胞和用于hpiv-1的ma104细胞中,用仅补充有谷氨酰胺的emem繁殖。在感染后3到4天收集含病毒培养物上清液,同时监测细胞病变效应,并通过离心(3,000rcf 15分钟)使其从细胞碎片中澄清。使用30kda amicon ultra过滤器单元将病毒浓缩至少10倍,用于血细胞凝集抑制(hi)测定。神经氨酸酶抑制(nl)测定使用经peg沉淀、然后如下所述纯化的病毒。将澄清的hpiv-3或hpiv-1上清液与peg6000(8%最终浓度)和naci(0.4m最终浓度)混合,然后在4℃轻轻搅拌下孵育过夜。通过在4℃以3,000rcf离心30min使peg6000/hpiv复合物沉淀。弃去上清液,并在4℃使用与初始病毒悬浮液体积的至少1:40相对应的一定体积的gnte缓冲液(200mm甘氨酸,200mm naci,20mm tris-hci,2mm edta,ph 7.4)使沉淀重悬过夜。通过上下移液,之后使用带有“紧”杵的杜恩斯匀浆器(douncer)机械破碎残留的病毒聚集体,使病毒悬浮液均质化。将hpiv-3或hpiv-1均质物加载于在gnte缓冲液中制备的30%-60%非线性蔗糖梯度的顶部上,并在4℃以100,000rcf离心2h 30min,无需制动减速。将病毒浓缩在30%-60%蔗糖界面,然后收集并储存在-4℃用于nl测定。
[0364]
hpiv hn抑制剂:
[0365]
将化合物以冻干粉末形式提供,然后溶解在无菌水或dmso中以生成10mm母液。将溶液声波处理15min以允许完全溶解。将母液储存在-20℃的琥珀色玻璃小瓶中,并在使用前于适当的缓冲液中新鲜稀释。
[0366]
血细胞凝集抑制测定:
[0367]
在u型底96孔板测定中以一式两份评估hn抑制剂。对于每种测试浓度(25μl/孔,1x最终),将化合物在pbs中稀释为4x溶液。将每种稀释物与4个血细胞凝集单位(hau)的hpiv-3或hpiv-1(25μl/孔,最终1hau)混合,并在室温孵育20min。将当量体积(50μl/孔)的1%人红细胞(h-rbc)添加至每个孔中。然后将板在室温(22-23℃)孵育1h,然后读取血细胞凝集程度。hi ic
50
被认为是与1hau的未处理病毒悬浮液相比将血细胞凝集素活性(凝集)降低50%的抑制剂浓度。
[0368]
神经氨酸酶抑制测定:
[0369]
制备纯化的hpiv-3或hpiv-1、抑制剂和mun,并在na反应缓冲液[naoac 50mm、caci
2 5mm、ph 4.6(hpiv3)或5.0(hpiv-1)]中稀释。最初测量采用不同hpiv-3或hpiv-1稀释度的神经氨酸酶测定以确定待用于测定中的最低病毒浓度。用足够的纯化病毒进行神经氨酸酶测定以获得最大荧光信号,该信号是实验背景的至少5倍,从而将被认为是统计学显著的。以一式三份进行神经氨酸酶抑制(nl)测定。对于每种测试浓度,将2μl纯化的hpiv和4μl 2.5x抑制剂溶液(最终1x)添加到每个孔中。将板在室温保持20min,然后将4μl的5mm 2
′‑
(4-甲基伞形酮基)-α-d-n-乙酰神经氨酸(mun)(最终2mm)添加到每个孔中,然后将板在37℃在搅动(1100rpm)下孵育30min。通过向每个孔中添加190μl甘氨酸缓冲液(甘氨酸0.25m,ph 10.4)来终止酶促反应。通过向病毒中添加mun来包括阴性对照,然后酶促反应在t=0分钟终止。用tecan infinite m200 pro测量相对荧光(rf)。通过背景扣除(阴性对照rf)处理数据,然后用graphpadprism分析以计算ic
50
值(非线性回归(曲线拟合)、剂量-反应-抑制、3或4参数逻辑)。与未处理的病毒悬浮液相比将神经氨酸酶活性(相对荧光)降低50%的抑制剂浓度被认为是nl ic
50
值。以一式三份进行所有测定。
[0370]
原位酶联免疫吸附测定(elisa):
[0371]
原位elisa是评价病毒生长抑制的有用技术。它在一个步骤中测量受感染的llc-mk2细胞单层细胞表面处hpiv-3hn的表达水平。表达水平与非固定化病毒感染和再感染靶细胞的能力直接相关。在基于细胞的测定中评估最佳抑制剂之前,可进行mtt测定以评价化合物的细胞毒性。在以200ffu/孔接种于96孔板中的汇合llc-mk2细胞单层上进行感染。在37℃以一式三份进行hpiv3毒株(c243、js或ci002)感染并持续1h,每15min轻轻搅动一次。将化合物以250mm到2.5nm的最终浓度稀释为10倍稀释系列。去除接种物并用100μl/孔每种相应的化合物稀释物替代。通过使用相同的实验条件,减去抑制剂,并入用于感染的阳性对照。将受感染的细胞单层在37℃、5%co2下保持36-40h,用于病毒增殖。将病毒灭活并通过直接添加100μl 7.4%甲醛/pbs将细胞固定。将板在室温维持15min,然后用pbs洗涤3次,持续5min,然后通过在37℃用0.3%h2o2/pbs处理30min,将内源性过氧化物酶灭活。洗涤细胞单层并将其与小鼠单克隆igg抗hpiv-3hn(fitzgerald,克隆编号m02122321,2.0mg/ml)在5%乳/pbs中以1μg/ml在37℃孵育1h。将各孔用0.02%tween20/pbs洗涤3次,持续5min。将于5%乳/pbs中以1:4000稀释的山羊抗小鼠-lgg(h l)-hrp缀合物(biorad,编号1706516)添加至各孔中并在37℃孵育1h。将细胞单层用0.02%tween20/pbs洗涤,然后用pbs冲洗两次。将bd opteia tmb底物添加到每个孔中,然后将板在37℃孵育。在3-5min后通过每孔添加50μl 0.6m h2so4来终止酶促反应。通过使用xmarktm微孔板吸光度分光光度计读取每个孔在450nm处的吸光度(od)来获得原始数据。通过从初始od读数中减去阴性对照(未感染细胞)od获得最终od,并用graphpad prism4分析数据以计算ic
50
值(非线性回归(曲线拟合)、剂量反应-抑制、3或4参数逻辑)。ic
50
值被认为是与未处理的受感染细胞单层相比将450nm处的吸光度降低50%的抑制剂浓度。
[0372]
可使用已公布的模型,在针对离体分化的人气道上皮(hae)细胞的hpiv-3抑制测定中测试本发明的化合物。简单地说,测试程序如下:如先前所述(muller等,2013),分离、培养和分化人气道上皮(hae)细胞。简单地说,将人鼻气道上皮细胞分离、扩增并接种在胶原包被的可渗透膜支持物(transwell)上。在细胞汇合后,去除顶端培养基,并将细胞在气液界面处维持大约4到6周,以允许上皮分化。将含有纤毛细胞的培养物经由顶端表面接种
每个transwell 400个病灶形成单位的hpiv-3,持续1小时。恰好在细胞已被病毒感染1h后,将不同浓度的测试化合物添加到hae顶端侧(20μl/transwell)。如先前所公布的(guillon等人,2014),在llc-mk2细胞中感染后1、3和6天,通过使用病灶形成测定的病毒滴定或原位elisa评估病毒载量减少。
[0373]
结构生物学
[0374]
重组hn表达和纯化
[0375]
使用杆状病毒表达系统(invitrogen,carlsbad,ca),基于经过大幅修改的文献程序来表达hn蛋白。因此,将用于蜜蜂蜂毒肽信号肽(hbm)的核苷酸序列添加到编码hn胞外结构域(氨基酸125到572)的序列的下游。该序列(hbm hn)经密码子优化以用于草地贪夜蛾(spodoptera frugiperda)细胞(sf9)中的表达,并通过dna2.0基因合成服务(gene synthesis service)(dna2.0,menlo park,ca)以命名为hbm-hnhpiv-3
opt
的基因直接订购。将hbm-hnhpiv-3
opt
通过pcr扩增并连接到载体中,该载体提供用于纯化和检测目的的额外的c末端6-组氨酸标签(his-tag)。
[0376]
根据制造商的说明进行含有hbm-hnhpiv-3
opt
的重组杆状病毒的生成和扩增。用高moi的hbm-hnhpiv-3
opt
杆状病毒感染在insect-xpress无蛋白昆虫细胞培养基(lonza)中培养的sf9细胞(invitrogen)。感染后四天,收集含有重组hn的上清液以产生最高的蛋白质表达。通过离心(3,000rcf 15min)澄清上清液以去除细胞碎片,然后依照制造商的方案在histrap excel 5ml柱(ge healthcare life sciences,buckinghamshire,england)上纯化。用500mm咪唑溶液洗脱重组hn,并评估收集的级分的神经氨酸酶酶促活性(参见上文)。汇集活性最高的级分并使用10kda amicon ultra过滤器单元(millipore)将其浓缩到最终体积为800μl。在4℃进行采用superdex 75凝胶过滤柱(ge healthcare)上的快速蛋白液相色谱(amersham biosciences)的额外纯化步骤,并用frac-920收集1ml级分。评估如通过在280nm处监测级分收集所确定的含蛋白级分的神经氨酸酶酶促活性并进行sds-page。将纯化并浓缩的重组hn蛋白质储存在4℃。
[0377]
结晶、数据收集和结构确定
[0378]
通过将晶体在含有5mm抑制剂的结晶溶液(0.1m柠檬酸盐缓冲液ph 4.6、0.2m(nh4)2so4、15%v/v聚乙二醇(peg)3000)中浸泡1hr-24hr的各种时间来制备一些hpiv3-hn复合物(与化合物ie-1826.23)。通过共结晶(与化合物ie-1826.30、ie-1530.74、ie-1530.69和ie-1778.39)制备其他hpiv-3hn复合物,其中将4mg/ml hpiv3 hn蛋白母液与最终浓度为1.5mm的抑制剂在0.1m柠檬酸盐缓冲液(ph 4.6)、0.2m(nh4)2so4和10%peg 3000中预孵育30min。使用悬滴蒸气扩散法将以2μl预孵育母液形式设立结晶试验。将液滴对500μl储液(reservoir)(0.1m柠檬酸盐缓冲液(ph 4.6)、0.2m(nh4)2so4和10%或15%peg 3000)进行平衡。将晶体安装在尼龙环(hampton research)中,并在除含沉淀剂溶液外还含20%甘油的冷冻保护剂溶液中在100k快速冷冻。
[0379]
使用blu-ice软件在澳大利亚同步加速器(australian synchrotron)的mx2光束线上收集x射线衍射数据。使用xds处理数据集,并使用ccp4套件中的aimless进行缩放。使用phaser和作为模板的apohpiv3-hn模型(pdb id:4xjq)通过分子置换来解析结构。使用phenix.refine改进模型,并使用molprobity进行结构验证。使用coot和pymol(http://www.pymol.org/;delano scientific llc)进行结构分析。
[0380]
结果
[0381]
表1:关于抑制剂实例对hpiv-3毒株c243的生物学评价.ni:神经氨酸酶抑制。
[0382]
[0383]
[0384]
[0385]
[0386][0387]
表2:关于hpiv-3(毒株c243、js、ci002)和hpiv-1(毒株c35)对抑制剂实例的生物学评价.hi:血细胞凝集抑制。ni:神经氨酸酶抑制。ic
50
:与实验阳性对照(不存在抑制剂)相比,将病毒功能/生长降低50%的抑制剂浓度。hpiv1毒株:c35。hpiv3毒株:c243、js、ci002(临床分离物)。
[0388]
[0389]
[0390]
[0391]
[0392]
[0393]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。