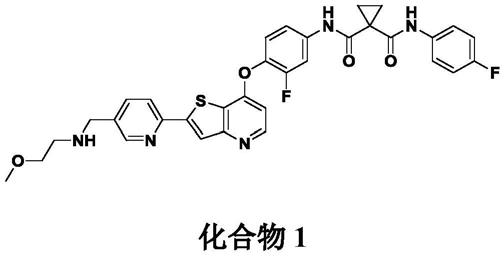
1.本发明涉及多酪氨酸激酶抑制剂的晶型。特别地,本发明涉及多酪氨酸激酶抑制剂n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的晶型、包含该晶型的药物组合物、制备该晶型的方法,及其使用方法。
背景技术:
[0002]
国际公开号wo2009/026717a公开具有多种蛋白酪氨酸激酶的抑制活性的化合物,例如vegf受体激酶和hgf受体激酶的抑制活性。特别地,公开的n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基))氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(化合物1)是一种多酪氨酸激酶抑制剂,表现出一系列密切相关的酪氨酸激酶的有效抑制,包括ret、cbl、chr4q12、ddr和trk,它们是导致细胞生长、存活和肿瘤进展的信号通路的关键调节剂。
[0003][0004]
化合物1在小鼠中的多个人异种移植肿瘤模型中显示肿瘤消退,并且目前在人临床试验中作为单一疗法以及联合治疗多种实体瘤。化合物1目前处于针对患有晚期癌症的患者的1期临床试验中,处于针对患有晚期脂肪肉瘤和非小细胞肺癌(nsclc)的患者的2期研究中。
[0005]
无定形的化合物1的小规模化学合成已在wo2009/026717a的实施例52(化合物147)中公开,然而,为了高质量并且大量地制备化合物1的api,通常将需要化合物1的晶型,从而可以通过重结晶清除工艺杂质。实际上,难以有把握预测特定化合物的哪种晶型将是稳定的、可重复的并适用于药物加工。甚至更难以预测是否会产生具有药物制剂所需物理性质的特定结晶固态形式。
[0006]
出于所有上述原因,非常需要生产提供药物组合物的制造改进的化合物1的晶型。本发明有利地解决这些需求中的一个或多个。
技术实现要素:
[0007]
本技术公开通过提供其晶型来解决上述挑战和需要的发明。本发明的发明人出乎意料地发现,对于n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n'-(4-氟苯基)环丙烷-1,1-二甲酰胺(以下称为化合物1),获
得单相晶型是非常困难的。在很努力地进行系统的固体形式筛选研究后,本发明的发明人获得化合物1的几种结晶材料,材料a至l,以及晶型c、d和h。然而,只有三种晶型(晶型c、d和h)以相均匀的形式存在,所有其他形式都由无序的多相组成。最后,本发明的发明人发现只有晶型d具有适用于药物制剂的优良物理性质,并且可以以高质量和良好的再现性以大规模商业规模生产。
[0008]
在本发明的第一方面,本文提供的是化合物1的晶型。
[0009]
在一个实施方案中,化合物1的晶型指定为晶型c、晶型d和晶型h。
[0010]
在一个实施方案中,化合物1的晶型指定为晶型d,其具有包含具有独立地选自7.8
±
0.2
°
、14.3
±
0.2
°
和17.2
±
0.2
°
的
°
2θ角值的衍射峰的x射线粉末衍射图(xrpd)。
[0011]
在一个实施方案中,化合物1的晶型(晶型d)可以是单晶。
[0012]
在一个实施方案中,化合物1的晶型指定为晶型c。
[0013]
在一个实施方案中,化合物1的晶型指定为晶型h。
[0014]
在另一个实施方案中,化合物1的晶型具有基本上如图1a、图2a(1)、图3b、图3c或图3d所示的xrpd图。
[0015]
在本发明的第二方面,提供用于所述方法中的药物组合物,所述药物组合物包含治疗有效量的化合物1的晶型,以及药学上可接受的赋形剂。在一个实施方案中,化合物1的晶型选自晶型c、晶型d和晶型h。
[0016]
在本发明的第三方面,本文提供用于抑制细胞中的多酪氨酸激酶活性的方法,所述方法包括使需要抑制多酪氨酸激酶活性的细胞与治疗有效量的化合物1的晶型接触。在一个实施方案中,化合物1的晶型选自晶型c、晶型d和晶型h。
[0017]
在本发明的第四方面,本文提供治疗有需要的受试者中的癌症的方法,所述方法包括向受试者施用治疗有效量的化合物1的晶型。在一个实施方案中,癌症为多酪氨酸激酶相关癌症。在一个实施方案中,多酪氨酸激酶相关癌症为肺癌,包括小细胞肺癌(nsclc)。在一个实施方案中,化合物1的晶型选自晶型c、晶型d和晶型h。
[0018]
在本发明的第五方面,本文提供用于制备化合物1的晶型的方法。在一个实施方案中,化合物1的晶型选自晶型c、晶型d和晶型h。
附图说明
[0019]
图1a说明根据实施例2a制备的化合物1的晶型d(化合物1晶型d)的x射线粉末衍射(xrpd)图。
[0020]
图1b说明根据实施例2a制备的化合物1的晶型d(化合物1晶型d)的差示扫描量热法(dsc)曲线。
[0021]
图2a:图2a(1)说明根据实施例2b制备的化合物1的晶型d(化合物1晶型d,单晶)的模拟x射线粉末衍射(xrpd)图;图2a(2)说明根据实施例2b制备的化合物1的晶型d(化合物1晶型d)的模拟(单晶)、实验xrpd图的叠加光谱。
[0022]
图2b说明根据实施例2b制备的化合物1的晶型d(化合物1晶型d,单晶)的晶体结构。
[0023]
图2c说明根据实施例2b制备的化合物1的晶型d(化合物1晶型d,单晶)的晶体包装。
[0024]
图3a说明实施例3中化合物1的晶型c、d和h(化合物1晶型c、d和h)的x射线粉末衍射(xrpd)图。
[0025]
图3b说明实施例3中化合物1的晶型c(化合物1晶型)的x射线粉末衍射(xrpd)图谱的索引溶液。
[0026]
图3c说明实施例3中化合物1的晶型d(化合物1晶型d)的x射线粉末衍射(xrpd)图的标定溶液。
[0027]
图3d说明实施例3中化合物1的晶型h(化合物1晶型h)的x射线粉末衍射(xrpd)图的索引溶液。
[0028]
图3e实施例3中化合物1的材料a至l(化合物1材料a至l)的x射线粉末衍射(xrpd)图。
[0029]
图3f说明实施例3中在丙酮中产生的化合物1的晶型d(化合物1晶型d)的tga和dsc热谱图。样品在约40℃下真空干燥约2.5小时。
[0030]
图3g说明实施例3中化合物1的结晶材料b(含有材料o)、o和a的x射线粉末衍射(xrpd)图。
具体实施方式
[0031]
本发明涉及化合物1的晶型。具体而言,本发明涉及选自化合物1的晶型c、晶型d或晶型h的晶型、包含该晶型的药物组合物、制备该晶型的方法及其使用方法。
[0032]
在一个实施方案中,化合物1的晶型指定为晶型c、晶型d和晶型h。
[0033]
在一个实施方案中,化合物1的晶型指定为晶型d,其具有包含具有独立地选自7.8
±
0.2
°
、14.3
±
0.2
°
和17.2
±
0.2
°
的
°
2θ角值的衍射峰的x射线粉末衍射图(xrpd)。
[0034]
在一个实施方案中,化合物1的晶型指定为晶型d,其具有包含具有独立地选自7.8
±
0.2
°
、14.3
±
0.2
°
、17.2
±
0.2
°
和21.7
±
0.2
°
的
°
2θ角值的衍射峰的x射线粉末衍射图(xrpd)。
[0035]
在一个实施方案中,化合物1的晶型指定为晶型d,其具有包含具有独立地选自7.8
±
0.2
°
、14.3
±
0.2
°
、17.2
±
0.2
°
、21.7
±
0.2
°
和26.4
±
0.2
°
的
°
2θ角值的衍射峰的x射线粉末衍射图(xrpd)。
[0036]
在另一个实施方案中,晶型d具有包含具有独立地选自7.8
±
0.2
°
、14.3
±
0.2
°
、16.6
±
0.2
°
、17.2
±
0.2
°
、19.3
±
0.2
°
、21.7
±
0.2
°
和26.4
±
0.2
°
的角度值的衍射峰的x射线粉末衍射图。
[0037]
在另一个实施方案中,晶型d具有包含具有独立地选自7.8
±
0.2
°
、14.3
±
0.2
°
、16.6
±
0.2
°
、17.2
±
0.2
°
、19.3
±
0.2
°
、21.7
±
0.2
°
、23.3
±
0.2
°
、26.4
±
0.2
°
和28.2
±
0.2
°
的角度值的衍射峰的x射线粉末衍射图。
[0038]
在另一个实施方案中,晶型d具有包含具有独立地选自6.50
±
0.2
°
、7.8
±
0.2
°
、14.3
±
0.2
°
、16.6
±
0.2
°
、17.2
±
0.2
°
、19.3
±
0.2
°
、21.7
±
0.2
°
、23.3
±
0.2
°
、25.7
±
0.2
°
、26.4
±
0.2
°
和28.2
±
0.2
°
的角度值的衍射峰的x射线粉末衍射图。
[0039]
在另一个实施方案中,晶型d具有基本上如图1a所示的xrpd图。
[0040]
在另一个实施方案中,晶型d的特征在于具有通过差示扫描量热法(dsc)测定的在大约57℃至62℃(起始~20℃至22℃)处具有峰值最大值的宽的小吸热线,随后是在大约
180℃(起始~178℃)处具有峰值最大值的急剧吸热。在另一个实施方案中,晶型d具有基本上如图1b所示的dsc热谱图。
[0041]
在一个实施方案中,化合物1的晶型d可以是单晶,其晶体结构数据总结在表1b中。如图2b所示,化合物1晶型d的单晶结构属于p-1空间群和三斜晶系。发现末端长烷基链具有大的椭圆体,表明与无序原子的高流动性。在晶体堆积的烷基链周围可以找到一些通道,见图2c。
[0042]
表1b.化合物1晶型d(单晶)的晶体数据和结构细化
[0043]
[0044]
[0045][0046]
在另一个实施方案中,晶型d具有基本上如图2a(1)所示的xrpd图。
[0047]
在一个实施方案中,晶型d具有基本上如图3c所示的xrpd图。在另一个实施方案中,晶型d具有基本上如图3f所示的dsc和/或tga。
[0048]
在一个实施方案中,化合物1的晶型被指定为晶形c。在另一个实施方案中,晶形c具有基本上如图3b所示的xrpd图。
[0049]
在一个实施方案中,化合物1的晶型指定为晶型h。在另一个实施方案中,晶型h具有基本上如图3d所示的xrpd图。
[0050]
在一个实施方案中,本发明的晶型是至少40%、50%、60%、70%、80%、90%或95%的纯化晶型。
[0051]
在本发明的第二方面,提供用于所述方法中的药物组合物,所述药物组合物包含治疗有效量的化合物1的晶型,以及药学上可接受的赋形剂。在一个实施方案中,化合物1的晶型选自晶型c、晶型d和晶型h。在另一个实施方案中,化合物1的晶型是晶型d。
[0052]
合物1的晶型可以通过本领域众所周知的任何方法配制,并且可以被制备成通过任何途径施用,包括但不限于肠胃外、口服、舌下、经皮、局部、鼻内、气管内或直肠内施用。在某些实施方案中,化合物1的晶型在医院场所中静脉内施用。在一个实施方案中,施用可以通过口服途径进行。
[0053]
载剂特征将取决于施用途径。如本文所使用的术语“药学上可接受的”表示与生物系统例如细胞、细胞培养物、组织或生物相容并且不干扰一种或多种活性成分的生物活性有效性的无毒材料。因此,除了抑制剂之外,组合物还可以含有稀释剂、填充剂、盐、缓冲剂、稳定剂、增溶剂和所属领域中熟知的其它材料。药学上可接受的制剂的制备描述于例如《雷氏药学大全(remington's pharmaceutical sciences)》,第18版,编者a.gennaro,宾夕法尼亚州伊斯顿的马克出版公司(mack publishing co.,easton,pa.),1990年中。
[0054]
在一个实施方案中,本发明的药物组合物含有95%的化合物1的晶型。在另一个实施方案中,本发明的药物组合物包含至少95%的化合物1的晶型。在另一个实施方案中,本发明的药物组合物含有至少90%的化合物1的晶型。在另一个实施方案中,本发明的药物组合物含有至少80%的化合物1的晶型。在另一个实施方案中,本发明的药物组合物含有至少70%的化合物1的晶型。在另一个实施方案中,本发明的药物组合物含有至少60%的化合物1的晶型。在另一个实施方案中,本发明的药物组合物含有至少50%的化合物1的晶型。
[0055]
包含化合物1的晶型的药物组合物可以用于本文所述的使用方法中。
[0056]
在本发明的第三方面,本文提供用于抑制细胞中的多酪氨酸激酶活性的方法,所述方法包括使需要抑制多酪氨酸激酶活性的细胞与治疗有效量的化合物1的晶型接触。在
一个实施方案中,化合物1的晶型选自晶型c、晶型d和晶型h。在另一个实施方案中,化合物1的晶型是晶型d。
[0057]
在本发明的第四方面,本文提供治疗有需要的受试者中的癌症的方法,所述方法包括向受试者施用治疗有效量的化合物1的晶型。
[0058]
本文提供的组合物和方法可用于治疗多种癌症。特定类型癌症的实例包括但不限于乳腺癌、肺癌,包括非小细胞肺癌(nsclc)、结肠癌、直肠癌、膀胱(尿路上皮)癌、前列腺癌、白血病、肾癌(肾)癌、神经胶质瘤、肉瘤,包括平滑肌肉瘤、肝(肝细胞)癌、卵巢癌和胃癌。
[0059]
在一个实施方案中,癌症为多酪氨酸激酶相关癌症。
[0060]
在一个实施方案中,多酪氨酸激酶相关癌症是nsclc、脂肪肉瘤、尿路上皮癌和口腔癌。在一个实施方案中,多酪氨酸激酶相关癌症为小细胞肺癌(nsclc)。
[0061]
活性化合物以足以向患者递送治疗有效量而不会在所治疗的患者中造成严重毒性作用的量包括于药学上可接受的载剂或稀释剂中。在一个实施方案中,对于所有上文所提到的条件,活性化合物的剂量范围为约0.6至1800mg/天,例如5至1000mg/天,并且再例如50至约200mg接受者/天。在一个实施方案中,活性化合物以150mg的剂量施用,优选口服,优选以连续21天的周期施用。合适载剂中的典型局部剂量范围将为0.01-3%wt/wt。药学上可接受的衍生物的有效剂量范围可以基于待递送的母化合物的重量来计算。如果衍生物本身展现活性,则可以如上文,使用衍生物的重量或通过所属领域的技术人员已知的其它手段估计有效剂量。
[0062]
在本文所描述的任何方法的一些实施方案中,在用本发明的组合物或方法治疗之前,用化学疗法、靶向抗癌剂、辐射疗法和手术中的一者或多者治疗患者,并且任选地,先前治疗不成功;和/或已经向患者施用手术,并且任选地,手术不成功;和/或已经用铂基化疗剂治疗患者,并且任选地,先前已经确定患者对用铂基化疗剂进行的治疗无反应;和/或已经用激酶抑制剂治疗患者,并且任选地,用激酶抑制剂进行的先前治疗不成功;和/或用一种或多种其它治疗剂治疗患者。
[0063]
在一个实施方案中,多酪氨酸激酶抑制剂每天口服施用一次。在一个实施方案中,化合物1的晶型每天口服施用两次。在一个实施方案中,化合物1的晶型每天口服施用一次。在一个实施方案中,化合物1的晶型选自晶型c、晶型d、晶型h。在另一个实施方案中,化合物1的晶型是晶型d。
[0064]
所属领域的技术人员应认识到,使用合适的、已知的和普遍接受的细胞和/或动物模型进行的活体内试验和活体外试验两者均预测所述组合的测试化合物或所述组合治疗或预防既定病症的能力。
[0065]
所属领域的技术人员将进一步认识到,可以根据临床和医学领域中熟知的方法在健康患者和/或患有既定病症的患者中完成人类临床试验,包括首次人类试验、剂量范围和功效试验。
[0066]
在本发明的第五方面,本文提供用于制备n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(化合物1)的晶型d的方法,包括以下任一程序:
[0067]
1)在丙酮中溶解n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻
吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,加热回流,搅拌冷却,以获得晶型d;
[0068]
2)在丙酮中溶解n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,晶型d,作为晶种,加热回流,搅拌冷却,以获得晶型d;
[0069]
3)在常温下在有机溶剂中研磨n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,过滤浆料,用新鲜有机溶剂代替有机溶剂,以获得晶型d,其中所述有机溶剂选自丙酮、acn、chcl3、mtbe、dmf、etoh、硝基甲烷或其混合物;
[0070]
4)在thf中溶解n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,蒸发,在etoac中溶解,沉淀,以获得晶型d;和,
[0071]
5)在约40℃下在ipa中研磨n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,过滤浆料,用新鲜的ipa代替ipa,以获得晶型d。
[0072]
在一个实施方案中,制备n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的晶型d的方法,包括以下任一程序:
[0073]
1)在丙酮中溶解n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,晶型d,作为晶种,加热回流,搅拌冷却,以获得晶型d,加热回流1~2小时,冷却至20
±
5℃的内部温度至少24h,过滤并用丙酮洗涤滤饼,在真空下在≤45℃下干燥,以获得晶型d;
[0074]
2)在室温下在有机溶剂中研磨n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,在约24小时后过滤浆料,用新鲜有机溶剂代替有机溶剂,以获得晶型d,其中所述有机溶剂选自丙酮、acn、chcl3、mtbe、dmf、etoh、硝基甲烷或其混合物;
[0075]
3)在thf中溶解n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,蒸发,在etoac中溶解,搅拌,沉淀,以获得晶型d;
[0076]
4)在约40℃下在ipa中研磨n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺,在约24小时后过滤浆料,用新鲜ipa代替ipa,以获得晶型d。
[0077]
在另一个实施方案中,有机溶剂选自chcl3/mtbe(50/50,v/v)和dmf/acn(30/70,v/v)。在另一个实施方案中,将所得浆料研磨至多~2.5周。
[0078]
定义
[0079]
除非另外定义,否则本文所使用的所有技术和科学术语皆具有与本发明所属领域的技术人员通常所理解的含义相同的含义。本文所提及的所有专利、专利申请和公开都通过引用的方式并入。
[0080]
如本文所用,“化合物1”是指n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡
啶-2-基)噻吩并[3,2-b))]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺。
[0081]
如本文所用,“化合物1的晶型”是指n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)的晶型)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的晶型。
[0082]
如果没有其他字符类型与该化合物相关,则为呈现独特结晶xrpd图案的材料指定连续的罗马字母字符作为默认名称。该名称暂时与术语“材料”相关联,直到通过xrpd图案的索引获得相纯度,并确定通过质子核磁共振光谱(1hnmr)获得的化学性质。当表征数据与由单相组成的独特晶型一致时,材料进一步指定为具有相同字母名称的“晶型”(即,材料c变为晶型c)。在本发明中,化合物的“晶型”的xrpd图可以成功地被索引。但是,“材料”的xrpd图案不能被索引,“材料”是具有某种程度无序的结晶材料,或是混合物。
[0083]
如本文所用,术语”晶型c”或”c晶型”单独使用时是指n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基))的c晶型甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的晶型。并且,术语“晶型d”或“d晶型”与“晶型c”或“c晶型”具有类似的含义。
[0084]
如本文所使用的“多酪氨酸激酶相关疾病或病症”是指与ret、cbl、chr4q12、ddr和/或trk中的致癌驱动突变相关或介导的疾病或病症。
[0085]
如本文所使用的可互换地使用的术语“受试者”、“个体”或“患者”是指任何动物,包括哺乳动物,例如小鼠、大鼠、其它啮齿动物、兔、狗、猫、猪、牛、绵羊、马、灵长类动物和人类。在一些实施方案中,患者为人类。在一些实施方案中,个体已经经历且/或展现待治疗和/或预防的疾病或病症的至少一种症状。在一些实施方案中,受试者疑似患有多酪氨酸激酶相关癌症。
[0086]
如本文所使用,化合物1的晶型的“治疗有效量”是足以改善、或以某种方式减轻症状,或停止或逆转病况的进展、或负调节或抑制多酪氨酸激酶活性的量。所述量可以以单一剂量形式施用或可以根据方案施用,由此其为有效的。
[0087]
如本文所使用的“治疗”表示其中病况、病症或疾病的症状或病理得到改善或另外得到有益地更改的任何方式。治疗还涵盖本文组合物的任何药学使用。
[0088]
如本文所使用的通过施用特定药物组合物来改善特定病症的症状是指可归因于组合物的施用或与组合物的施用相关的任何减轻,无论是永久的还是暂时的、持续的还是短暂的减轻。
[0089]
如本文所使用的术语“约”当用于修饰数值上定义的参数(例如本文详述的化合物1的晶型或其药学上可接受的盐的剂量,或本文所述的治疗时间的时长)时表示所述参数的变化程度可能会比针对该参数的陈述数值低或高多10%。举例来说,约5mg/kg的剂量可以在4.5mg/kg与5.5mg/kg之间变化。“约”在参数清单开头使用时旨在修饰每个参数。举例来说,约0.5mg、0.75mg或1.0mg表示约0.5mg、约0.75mg或约1.0mg。同样地,约5%或更多、10%或更多、15%或更多、20%或更多和25%或更多表示约5%或更多、约10%或更多、约15%或更多、约20%或更多和约25%或更多。
[0090]
如本文所用,术语“约”在用于指xrpd峰位置时是指峰的固有可变性,这取决于仪器的校准、用于制备本发明的晶型的方法、晶型的年龄和分析中使用的仪器类型。用于xrpd分析的仪器的可变性约为
±
0.2
°ꢀ
2θ。
[0091]
如本文所用,术语“约”在用于指代dsc吸热峰开始时是指峰的固有可变性,这取决于仪器的校准、用于制备本发明样品的方法以及所用仪器的类型在分析中。用于dsc分析的仪器的可变性约为
±
2℃。
[0092]
通用方法
[0093]
除非另有说明,否则下文概述的一般方法用于示例性实施例中。
[0094]
i.结晶技术
[0095]
本发明的晶型可以使用本领域技术人员熟知的多种方法制备,包括从合适的溶剂中结晶或重结晶或通过升华。可以使用多种技术,包括示例性实施例中的那些技术,用于结晶或重结晶,包括蒸发与水混溶或与水不混溶的溶剂,在过饱和溶剂混合物中晶种,降低溶剂混合物的温度,或冷冻干燥溶剂混合物。
[0096]
在本发明中,结晶可以在有或没有晶种的情况下进行。晶种可以来自任何先前批次的所需晶型。晶种的加入不会影响本发明晶型的制备。
[0097]
在等温线完成后回收样品并通过xrpd重新分析。
[0098]
缩写和首字母缩略词
[0099]
[0100][0101]
以下实施例旨在进一步说明本发明的某些实施方案,而不旨在限制本发明的范围。
[0102]
实施例1
[0103]
n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)的制备))苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(化合物1)
[0104]
本实施例说明n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(化合物1)的制备。
[0105]
步骤1:n-((6-溴代吡啶-3-基)甲基)-2-甲氧基乙-1-胺(化合物1a)
[0106][0107]
向搅拌的2-甲氧基乙胺(3.0当量)在二氯甲烷(dcm)(12体积)中的溶液中加入分子筛(0.3w/w)并在氮气氛下在25
±
5℃下搅拌2小时。通过karl fischer分析监测反应物质的水含量,直到水含量限值达到0.5%w/w。一旦达到水含量极限,将反应物质冷却至5
±
5℃,并在30min内将6-溴烟醛(1.0当量)分批加入5
±
5℃的上述反应物质中。将反应物质在5
±
5℃下搅拌30
±
5min并在5
±
5℃逐滴加入乙酸(1.05当量)。在加入完成后,将物质缓慢升温至25
±
5℃并搅拌8小时,以得到化合物1a。通过hplc监测亚胺形成。
[0108]
步骤2:((6-溴代吡啶-3-基)甲基)-(2-甲氧基乙基)-氨基甲酸叔丁基酯(化合物1b)
[0109][0110]
加入在thf(5.0体积)中的带电化合物1a(1.0当量)并将反应物质在氮气氛下在25
±
5℃下搅拌30min。将反应物质冷却至约10
±
5℃的温度。在氮气气氛下,在10
±
5℃下将二碳酸二叔丁酯(1.2当量)加到反应物质中,并将反应物质温度升至25
±
5℃,并且物质反应约2小时。通过hplc监测反应进程。在ipc完成后,加入在2m naoh(3.1体积)中制备的牛磺酸(1.5当量)溶液并在10
±
5℃下搅拌16h至18h。用1m naoh水溶液(3.7体积)进一步稀释反应物质并分离各层。水层用dcm(2 x 4.7体积)萃取并且提取物与有机层组合。将合并的有机层用1m naoh水溶液(3.94体积)洗涤,然后用水(2 x 4.4体积)洗涤,并且经硫酸钠(2.0w/w)干燥。在40℃下减压浓缩滤液,直到没有观察到滤液。依次加入四氢呋喃(thf)(1 x 4体积和1 x 6体积)并在低于40℃的减压下浓缩直到观察不到馏出物以获得化合物1b,为淡黄色浆料。
[0111]
步骤3:((6-(7-氯代噻吩并[3,2-b]吡啶-2-基)吡啶-3-基)甲基)(2-甲氧基乙基)氨基甲酸叔丁基酯(化合物1c)
[0112][0113]
在-15
±
10℃下,向搅拌的7-氯代噻吩并[3,2-b]吡啶(1.05当量)的四氢呋喃(7体积)溶液中逐滴加入正丁基锂(2.5m的己烷溶液)并在相同温度下在氮气气氛下搅拌90分钟。在-15
±
10℃下将氯化锌(1.05当量)加入反应物质中。将反应物质缓慢升温至25
±
5℃并在氮气氛下搅拌45min以提供化合物1c。通过hplc监测反应进程。
[0114]
步骤4:((6-(7-(4-氨基-2-氟苯氧基)噻吩并[3,2-b]吡啶-2-基)吡啶-3-基)甲基)(2-甲氧基乙基)氨基甲酸叔丁基酯(化合物1d)
[0115][0116]
在氮气气氛下,在25
±
5℃下加入dmso(3.9体积)中的3-氟-4-羟基苯胺氯化物(1.2当量),并搅拌反应物质直到在25
±
5℃下观察到澄清溶液。t-buok在25
±
10℃的氮气气氛下分批加入。将反应物质温度升至45
±
5℃并在氮气氛下保持30min。将化合物1c在45
±
5℃下在氮气氛下分批加入并在45
±
5℃下搅拌10分钟。将反应混合物加热至100
±
5℃并搅拌2小时。通过hplc监测反应物质。
[0117]
在反应完成后,将反应物质冷却至10
±
5℃并用10
±
5℃的冷水(20体积)淬灭。将
物质温度升至25
±
5℃并搅拌7-8小时。通过过滤收集所得化合物1d粗品并用2体积水洗涤。将粗化合物1d材料置于水(10体积)中并在25
±
5℃下搅拌至多20min。将反应物质加热至45
±
5℃并在45
±
5℃下搅拌2-3h,过滤并真空干燥。
[0118]
将粗化合物1d在25
±
5℃下加到mtbe(5体积)中并在25
±
5℃下搅拌约20分钟。将反应物质温度升至45
±
5℃,在45
±
5℃下搅拌3-4小时,然后冷却至20
±
5℃。将反应物质在20
±
5℃下搅拌约20分钟,过滤,然后用水(0.5体积)洗涤并真空干燥。
[0119]
在25
±
5℃下将粗物质溶解在丙酮(10体积)中并在25
±
5℃下搅拌约2h。通过硅藻土床过滤反应物质并用丙酮(2.5vol)洗涤。滤液在25
±
5℃下用水(15体积)缓慢稀释。将反应物质在25
±
5℃下搅拌2-3h,过滤并用水(2体积)洗涤床并真空干燥,以得到化合物1d,为棕色固体。
[0120]
步骤5:1-((4-((2-(5-(((叔丁氧基羰基)(2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-))基)氧基)-3-氟苯基)氨基甲酰基)环丙烷-1-羧酸(化合物1e)
[0121][0122]
向化合物1d(1.0当量)的四氢呋喃(7体积)溶液中,加入碳酸钾水溶液(1.0当量)的水(8体积)溶液。将溶液冷却至5
±
5℃,并且搅拌约60min。在搅拌的同时,在5
±
5℃下将三乙胺(2.0当量)分别加到1,1-环丙烷二甲酸(2.0当量)的四氢呋喃(8体积)溶液中,然后加入亚硫酰氯(2.0当量)并搅拌约60min。在5
±
5℃下将酰氯物质缓慢加到化合物1d溶液中。将温度升至25
±
5℃并搅拌3.0小时。通过hplc分析监测反应。
[0123]
在反应完成后,用乙酸乙酯(5.8体积)、水(5.1体积)、10%(w/w)盐酸水溶液(0.8体积)和25%(w/w)氯化钠水溶液(2体积)稀释该物质。分离水层并用乙酸乙酯(2
×
5体积)萃取。用0.5m碳酸氢钠水溶液(7.5体积)洗涤合并的有机层。有机层用darco活性炭(0.5w/w)和硫酸钠(0.3w/w)在25
±
5℃下处理1.0h。有机层通过硅藻土过滤并用四氢呋喃(5.0体积)洗涤。将滤液在低于50℃的真空下浓缩至约3体积,并在低于50℃的真空下与乙酸乙酯(2x 5体积)共蒸馏至约3.0体积。将有机层冷却至15
±
5℃,搅拌约60min,过滤,并用乙酸乙酯(2.0体积)洗涤固体。将该物质在40
±
5℃下真空干燥直到水含量小于1%,以得到化合物1e,为棕色固体。
[0124]
步骤6:((6-(7-(2-氟-4-(1-((4-氟苯基)氨基甲酰基)环丙烷-1-甲酰胺基)苯氧基)噻吩并[3,2-b]吡啶-2-基))吡啶-3-基)甲基)(2-甲氧基乙基)氨基甲酸叔丁基酯(化合物1f)
[0125][0126]
将吡啶(1.1当量)加到化合物1e(1.0当量)在四氢呋喃(10体积)中的悬浮液中并冷却至5
±
5℃。加入亚硫酰氯(2.0当量)并搅拌约60min。将样品在甲醇中淬灭后,通过hplc分析确认生成的酰氯形成。另外,将碳酸钾(2.5当量)水溶液(7.0体积水)加到4-氟苯胺(3.5当量)的四氢呋喃(10体积)溶液中,冷却至5
±
5℃,并搅拌约60min。将5
±
5℃的酰氯物质的温度升至约25
±
5℃的温度并搅拌3h。通过hplc分析监测反应。
[0127]
在反应完成后,溶液用乙酸乙酯(25体积)稀释,分离有机层并用1m氢氧化钠水溶液(7.5体积)、1m盐酸水溶液(7.5体积)洗涤,和25%(w/w)氯化钠水溶液(7.5体积)洗涤。有机层经硫酸钠干燥并过滤(1.0w/w)。在低于50℃的真空下将滤液浓缩~3体积,并在低于50℃的真空下与乙酸乙酯(3 x 5体积)共蒸馏至~3.0体积。加入乙酸乙酯(5体积)和mtbe(10体积),加热至50
±
5℃并搅拌30-60min。将混合物冷却至15
±
5℃,搅拌约30min,过滤,并用乙酸乙酯(2.0体积)洗涤固体。通过hplc分析分析mgb3含量。将该物质在40
±
5℃下真空干燥直到水含量达到约3.0%,以得到化合物1f,为棕色固体。
[0128]
步骤7:n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(化合物1)
[0129][0130]
向化合物1f在冰乙酸(3.5体积)的混合物中加入浓盐酸(0.5体积)并在25
±
5℃下搅拌1.0h。通过hplc分析监测反应。
[0131]
在反应完成后,将物质加到水(11体积)中并在20
±
5℃下搅拌30min。使用10%(w/w)碳酸氢钠水溶液将ph值调节至3.0
±
0.5,并在20
±
5℃下搅拌约3.0h。过滤物质,用水(4 x 5.0体积)洗涤,每次洗涤后检查滤液的ph值。将该材料在50
±
5℃的真空下干燥直到水含量为约10%。
[0132]
将粗化合物1置于乙酸乙酯(30体积)中,加热至70
±
10℃,搅拌1.0小时,冷却至25
±
5℃,过滤,并用乙酸乙酯(2体积)洗涤。将该材料在真空下在45
±
5℃下干燥6.0h。
[0133]
将粗化合物1放入经过抛光过滤的四氢呋喃(30体积)和预洗涤的amberlyst a-21离子交换树脂中,并在25
±
5℃下搅拌直到溶液变得澄清。在得到澄清溶液后,过滤树脂并用抛光过滤的四氢呋喃(15体积)洗涤。滤液在低于50℃的真空下浓缩约50%,并与精制过滤的ipa(3 x 15.0体积)共蒸馏,并在低于50℃的真空下浓缩至约50%。加入带电抛光过滤
的ipa(15体积)并将溶液在低于50℃的真空下浓缩至~20体积。将反应物质加热至80
±
5℃,搅拌60min。并冷却至25
±
5℃。将所得反应物质在25
±
5℃下搅拌约20小时。将反应物质冷却至0
±
5℃,搅拌4-5小时,过滤,并用抛光过滤的ipa(2体积)洗涤。将物质在45
±
5℃下真空干燥至含水量约2%,得到所需产物化合物1。1h-nmr(400mhz,dmso-d6):δ10.40(s,1h),10.01(s,1h),8.59
–
8.55(m,1h),8.53(d,j=5.6hz,1h),8.32(s,1h),8.23(d,j=8.0hz,1h),7.96
–
7.86(m,2h),7.70
–
7.60(m,2h),7.56
–
7.43(m,2h),7.20
–
7.11(m,2h),6.66(d,j=5.6hz,1h),3.78(s,2h),3.41(t,j=5.6hz,2h),3.25(s,3h),2.66(t,j=5.6hz,2h),1.48(s,4h)ppm。ms:m/e 630(m 1)
。
[0134]
实施例2
[0135]
n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺的晶型d的制备
[0136]
实施例2a:化合物1晶型d的制备
[0137]
向50l反应器中加入7.15kg化合物1、40g作为晶种的晶型d和21l丙酮(≥99%)。经1~2h将混合物加热至回流(~56℃)。将混合物在20
±
5℃的内部温度的情况下搅拌至少24h。然后,过滤悬浮液并用7l丙酮洗涤滤饼。湿饼在45℃下在真空下干燥≤,以获得5.33kg所需晶型d的化合物1。
[0138]
x射线粉末衍射(xrpd)
[0139]
xrpd图案是使用pan alytical x'pert pro mpd衍射仪收集的,该衍射仪使用使用au optix长细聚焦源产生的cu辐射的入射光束。椭圆渐变多层镜用于聚焦穿过样品和在探测器上的cu ka x射线。在分析之前,对硅样品(nist srm 640e)进行分析以验证观察到的si iii峰位置与nist认证的位置一致。每个样品的样本夹在3-μm厚的薄膜之间,并进行透射几何分析。光束停止器、短自散射扩展和自散射刀刃用于最小化由空气产生的背景。入射和衍射束的索勒狭缝用于最大限度地减少轴向发散的展宽。衍射图案使用位于样本的240mm的扫描位置敏感检测器(x'celerator)和数据收集器软件v.2.2b收集。pattern match v2.3.6用于形成xrpd图。
[0140]
x射线粉末衍射(xrpd)图用于表征获得的化合物1,表明化合物1为化合物1的晶型d(化合物1晶型d),见图1a。产生的xrpd图与图3c中所示的基本相同。
[0141]
差示扫描量热法(dsc)
[0142]
使用mettler-toledo dsc3 差示扫描量热仪进行dsc。使用辛烷、水杨酸苯酯、铟、锡和锌进行温度校准。tawn灵敏度为11.9。将样品放入铝制dsc盘中,盖上盖子,准确记录重量。将配置为样品盘的称重铝盘放置在池的参考侧。在样品分析之前,盘盖被刺穿。热谱图上的方法名称是开始和结束温度以及加热速率的缩写;例如,-30-250-10表示“在10℃/min下环境温度至250℃”。氮气流速为50.0ml/min。本仪器不提供由usp要求的气压值,因为它与大气压相同。
[0143]
观测到在大约57℃至62℃(开始~20℃至22℃)处具有峰最大值的宽的小吸热,然后是在大约180℃(开始~178℃)处具有峰最大值的急剧吸热。这些事件可能分别是由于挥发物的损失和熔体(参见图1b)。
[0144]
在一个备选实施方案中,如下制备晶型d。将指定材料o悬浮在600μl丙酮中。观察到初始溶解,然后再沉淀。未测量悬浮固体的量,因为该实验的目标是获得具有足够固体的
悬浮液以浆化、分离和收集xrpd数据。根据晶型d在丙酮中的溶解度,对实验规模的粗略估计约为80-100mg。将悬浮液在环境温度下搅拌约2.5周,然后通过离心过滤分离固体。xrpd数据似乎与晶型d一致。然后将样品在真空烘箱中在约40℃下干燥约2.5小时。最终固体的xrpd图与晶型d一致。
[0145]
实施例2b:化合物1晶型d的制备
[0146]
将427.0mg化合物1溶解在5ml thf中,得到澄清的棕色溶液。过滤所得溶液,滤液在氮气流下蒸发。获得粘性固体,将其在室温下真空干燥约5min,仍然获得粘性棕色固体。将其溶解在0.2ml的etoac中并超声溶解。将获得的溶液在室温下搅拌15min并且稀释固体。将所得固体加入0.4ml etoac并在室温下搅拌21h 40min以形成悬浮液。通过离心从母液中分离出固体,然后将所得固体重新悬浮在0.6ml etoac中并在室温下搅拌2天。通过离心分离固体,得到所需晶型d的化合物1。
[0147]
使用x射线粉末衍射(xrpd)图表征获得的化合物1,表明化合物1为化合物1的晶型d(化合物1晶型d)。
[0148]
实施例2c:化合物1晶型d的制备
[0149]
化合物1的单晶x射线衍射数据在rigaku xtalab pro 007hf(mo)衍射仪上以180k收集,具有mo kα辐射使用crysalispro程序进行数据缩减和经验吸收校正。该结构通过使用shelxt程序的双空间算法求解。所有非氢原子都可以直接从差分傅立叶图中定位。框架氢原子被几何放置并使用骑乘模型约束到母原子。最终的结构细化是使用shelxl程序通过使用全矩阵技术最小化f2的平方偏差之和来完成的。
[0150]
晶型d化合物1(单晶)的制备
[0151]
将化合物1晶型d溶解在丙酮/acn(1/2)的混合物中,其中化合物1的浓度为~7mg/ml。获得块状单晶,为单晶。
[0152]
xrpd图用于表征获得的晶型d化合物1的单晶,参见图2a。晶体结构数据在表1b中总结。细化的单晶结构如图2b所示。单晶结构化合物1晶型d属于p-1空间群和三斜晶系。发现末端长烷基链具有大的椭圆体,表明具有无序原子的高迁移率。
[0153]
从单晶结构计算的理论xrpd和实验xrpd基本类似(图2a)。由于取向偏好、无序和测试温度(单晶数据为180k,实验数据为293k),一些小峰不存在或发生偏移.
[0154]
表1b.化合物1晶型d(单晶)的晶体数据和结构细化
[0155]
[0156][0157]
实施例3
[0158]
n-(3-氟-4-((2-(5-(((2-甲氧基乙基)氨基)甲基)吡啶-2-基)噻吩并[3,2-b]吡啶-7-基)氧基)苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(化合物1)的固体筛选
[0159]
进行化合物1的固体形式筛选。当以足够量分离时,固体通过x射线粉末衍射(xrpd)图案进行表征。将xrpd模式与各自进行比较。此外,其他光谱或方法,例如差示扫描量热法(dsc)、热重分析法(tga)和动态蒸气吸附法(dvs),用于表征或研究获得的固体。
[0160]
x射线粉末衍射(xrpd)
[0161]
图案匹配版本2.3.6和3.0.4用于形成xrpd图覆盖。标有“patternmatch v3.0.4的图像”的图是使用未经验证的软件版本生成的,因此被视为非cgmp表示。
[0162]
1)传输模式xrpd
[0163]
xrpd图案是用帕纳科x'pert pro mpd衍射仪收集的,该衍射仪使用使用optix长细聚焦源产生的铜辐射入射光束。椭圆渐变多层镜用于将cu kαx射线辐射聚焦通过样品并到达检测器。在分析之前,对硅样品(nist srm 640d)进行分析以验证观察到的si(111)峰的位置与nist认证的位置一致。将样品样品夹在3μm厚的薄膜之间,并进行透射几何分析。使用光束停止、短抗散射延伸和抗散射刀刃来最小化空气产生的背景。用于入射光束和衍射光束的索勒狭缝用于最小化轴向发散引起的展宽。衍射图案使用距离样本240mm的扫描位置敏感探测器(x'celerator)和data collector软件v.2.2b.收集。
[0164]
2)反射模式xrpd
[0165]
vt-xrpd图案是用帕纳科x'pert pro mpd衍射仪收集的,使用的入射光束是使用长细聚焦源和镍滤光片产生的cu kα辐射。使用对称bragg
–
brentano几何配置衍射仪。数据使用data collector软件v.2.2b收集和分析。在分析之前,对硅样品(nist srm 640d)进行分析以验证观察到的si(111)峰的位置与nist认证的位置一致。将样品的样品装入镀镍铜孔中。抗散射狭缝(ss)用于最小化空气产生的背景。用于入射光束和衍射光束的索勒狭缝用于最小化轴向发散引起的展宽。衍射图案使用距离样品240mm的扫描位置敏感检测器(x'celerator)和data collector软件v.2.2b收集。
[0166]
3)可变温度xrpd
[0167]
anton paar温湿度室(thc)用于收集随温度变化的原位反射模式xrpd图案。使用位于样品架正下方的peltier热电设备改变样品的温度,并使用位于样品架中的铂100电阻传感器进行监测。热电设备由与数据收集器连接的anton paar tcu 50供电和控制。
[0168]
偏光显微镜(plm)
[0169]
使用带有一级红色补偿器的leica mz12.5立体显微镜观察样品。通常使用范围为0.8-10倍的各种物镜与交叉偏振光一起观察样品。在矿物油中观察到选定的样品。使用配备spot insight彩色数码相机的leica dm lp显微镜分析选定的样品。将每个样品置于载玻片上,在样品上放置盖玻片,并加入一滴矿物油以通过毛细作用覆盖样品。使用带有交叉偏振器和一阶红色补偿器的20 x 0.40na和40 x 0.74na放大倍率观察每个样品。图像使用spot软件(v.4.5.9)捕获。在每个图像上插入微米条作为粒度的参考。
[0170]
差示扫描量热法(dsc)
[0171]
dsc分析使用ta instruments 2920和q2000差示扫描量热仪进行。温度校准使用nist可追溯的铟金属进行。将样品放入铝制dsc盘中,盖上盖子,准确记录重量。将配置为样品盘的称重铝盘t0c或t0chsmp(分别用手动针孔盘tzero卷边或tzero气密密封)放置在池的参考侧。热谱图上的方法代码是开始和结束温度以及升温速率的缩写;例如,-30-250-10表示“在10℃/min下-30℃至250℃。”[0172]
热重法(tga)
[0173]
tg分析使用ta instruments 2950热重分析仪进行。使用镍和alumel
tm
进行温度校准。将每个样品置于铝盘中并插入tg炉中。在氮气吹扫下加热炉子。热谱图上的方法代码是
开始和结束温度以及升温速率的缩写;例如,25-350-10表示“在10℃/min下25℃至350℃”。
[0174]
动态蒸汽吸附(dvs)
[0175]
在vti sga-100蒸汽吸附分析仪上收集自动蒸汽吸附(vs)数据。nacl和pvp用作校准标准。样品在分析前未干燥。在氮气吹扫下以10%rh增量在5%至95%rh范围内收集吸附和解吸数据。用于分析的平衡标准是5分钟内重量变化小于0.0100%,最大平衡时间为3小时。数据未针对样品的初始水分含量进行校正。
[0176]
索引
[0177]
计算研究,即索引和结构细化在“ssci非cgmp活动程序”下进行。允许的峰位置和观察到的峰之间的一致性表明一致的晶胞测定。使用x'pert high score plus 2.2a(2.2.1)和专有的ssci软件进行索引。没有尝试进行分子填充来确认本工作范围内的暂定索引解决方案。
[0178]
实施例3a:材料a、材料b(含有材料o)、化合物1的材料o的制备
[0179]
材料a、b(包含观察到的化合物1的材料o和材料o)的xrpd图在图3g中给出。
[0180]
实施例3b:化合物1的稳定形式筛选
[0181]
将实施例3a中获得的化合物1的材料b(含有材料o)在包括具有高水活度的溶剂系统的各种溶剂中研磨以靶向潜在的水合物。实验大多在环境温度下进行。选定的实验在~40℃下进行,以达到足够的溶解度。为了改变化合物1的杂质分布,在大约24小时后过滤大部分浆料并用新鲜溶剂替换溶剂(如果需要,调整比率)。将所得浆料研磨至多约2.5周。稳定形式筛选的条件和结果总结在表2a中。
[0182]
表2a.化合物1的稳定形态筛选
[0183]
[0184][0185]
(a)除非另有说明,否则用溶剂代替进行研磨约2.5周。
[0186]
(b)在加入溶剂时观察到溶解,然后是沉淀。
[0187]
(c)研磨进行约2周,无需更换溶剂。
[0188]
(d)使用母液溶液通过重量法测定溶解度。
[0189]
(e)样品以薄膜、油或玻璃的形式出现。
[0190]
实施例3c:化合物1的多晶型筛选(溶液方法)
[0191]
化合物1的实施例3a中获得的材料b(含有材料o)经受各种溶液结晶技术,例如蒸发、蒸气扩散、冷却至环境或低于环境温度、浆料以及旨在形成亚稳态形式的溶剂/反溶剂沉淀。使用非水溶剂和具有高水活性的溶剂系统。多晶型物筛选的条件和结果(溶液实验)总结在表2b中。
[0192]
表2b.化合物1的多晶型筛选(溶液方法)
[0193]
[0194]
[0195][0196][0197]
(a)溶剂比、温度和实验持续时间是近似的。
[0198]
材料j是在选定的水性和非水性条件下生产的。材料j是结晶的。在约40℃的短期真空干燥导致存在另一种材料(称为材料m)。
[0199]
实施例3c:化合物1晶型的干燥实验
[0200]
有限的基于非溶剂的实验是用在稳定形式筛选过程中产生的材料进行的,包括在高温下的短期真空干燥。干燥实验的条件和结果总结在表2c中。
[0201]
表2c.基于无溶剂的实验
[0202]
起始材料条件xrpd结果晶型c真空烘箱干燥,~40℃,2.5小时晶型c晶型d真空烘箱干燥,~40℃,2.5小时晶型d晶型h真空烘箱干燥,~40℃,2.5小时晶型h材料j真空烘箱干燥,~40℃,2.5小时材料j m
[0203]
实施例3d:化合物1的固体形式的x射线粉末衍射(xrpd)图
[0204]
生产在实施例3a至3e中获得的几种材料,具有独特的xrpd图案。独特的材料被指定为晶型c、d、h和材料a、b、e、f、g、j、k、l、n和o。这些形式的xrpd图如图3a所示。观察到的其他材料的图案在图3b中给出。
[0205]
对晶型c(来自二噁烷)、晶型d和晶型h的xrpd图的进一步成功标引表明晶型c、d和h由单晶相组成。分别参见图3b、图3c和图3d。并且,在丙酮中产生的晶型d的tga和dsc热分析图如图3f所示。
[0206]
材料a至l的xrpd图无法索引,表明材料a到l是具有某种程度无序或混合物的结晶材料。参见图3e。其中,材料e是在异丙醇中进行的单一实验中观察到的,并且样品通过冷却制成并且可能含有另一种材料,称为材料g;材料f以中等和高水分活度生产,通常与另一种称为材料g的材料混合;作为与材料f的混合物观察到的材料g仅在具有较高水活度的溶剂系统中生产;材料k具有与材料g类似的xrpd图案。
[0207]
实施例2e:浆料相互转化实验
[0208]
为了研究筛选期间产生的所选材料之间的关系,进行有限的浆料相互转化实验。通过在环境温度下将感兴趣的固体种子在两种不同的溶剂系统中浆化来进行实验。所有材料在浆化前真空干燥(~40℃,~2.5小时)。选择溶剂条件以实现测试材料的足够溶解度。使用非水条件和具有高水活性的溶剂系统。结果总结在表2d中。
[0209]
将晶型c、d和h的浆料在乙酸乙酯中进行,结果是不确定的。该实验产生晶型c和晶型d的混合物,表明晶型h在这些条件下的热力学稳定性最低,但c和d的相对稳定性仍不清楚。混合物的xrpd峰强度的比较表明晶型c可能过量。然而,这可能是由于晶型h选择性地变成晶型c所致。因此,晶型c和晶型d的相对稳定性是未知的。材料f、g和j的种子在四氢呋喃/水(40/60)中的浆料产生材料f。
[0210]
表2d.浆料相互转化实验
[0211][0212]
(a)用真空干燥的起始材料(~40℃,~2.5小时)进行浆料处理约42小时。
[0213]
(b)将固体在被浆料饱和的溶剂中搅拌~3小时,用乙酸乙酯溶液代替溶剂。
[0214]
实施例2h:晶型d的初步物理稳定性评估
[0215]
对晶型d进行初步物理稳定性评估。数据总结在表2e中。
[0216]
实验表明晶型d在承受约43%和约75%rh应力以及在约40℃下进行短期真空干燥时保持不变。与未经处理的样品相比,受应力材料的xrpd图没有显示出显著的峰位移。从高水分活度浆料获得的晶型d的xrpd图案与未处理材料的图案一致。然而,观察到所选峰的微小变化,可能是由于水合。
[0217]
表2e.晶型d的初步稳定性评估
[0218]
条件(a)xrpd结果~43%rh/rt与晶型d一致,峰无明显变化~75%rh/rt与晶型d一致,峰无明显变化丙酮/水(40/60),浆料,rt与晶型d一致,峰有轻微偏移etoh/水(40/60),浆料,rt与晶型d一致,峰有轻微偏移水,浆料,rt与晶型d一致,峰有轻微偏移
[0219]
虽然已经结合本发明的特定实施例描述了本发明,但应理解,其能够进一步修改,并且本技术意图涵盖本发明的任何改变、使用或调适,一般来说,遵循本发明的原理并且包括与本公开的所述偏离,如在本发明所涉及的领域内的已知或惯用实践内并且如可以应用于上文所阐述的基本特点并且如下在所附权利要求书的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。