1.本发明涉及药物化学领域,以及该合成方法的新中间体及其合成方法。
背景技术:
2.2-(溴甲基)-2-丁基己酸(英文名:2-(bromomethyl)-2-butylhexanoic acid),专利cn113677398a(阿尔比里奥公司)报道发现2-(溴甲基)-2-丁基己酸是合成1,2,5-苯并硫杂二氮杂环庚三烯衍生物的关键中间体。1,2,5-苯并硫杂二氮杂环庚三烯衍生物为具有顶端钠依赖性胆汁酸转运蛋白(asbt)及/或肝脏胆汁酸转运(lbat)抑制活性的胆汁酸调节剂。这些化合物潜在可用于治疗心血管疾病、脂肪酸代谢及葡萄糖利用病症、胃肠道疾病及肝病。
3.cn106573902a(cj医药健康株式会社)公开了一种新型氨基烷基苯并硫氮杂衍生物及其用途,该新型氨基烷基苯并硫氮杂衍生物作为活性成分的可用于预防或治疗便秘。
4.然而作为该类药物活成成分的关键中间体2-(溴甲基)-2-丁基己酸也备受关注,发现2-(溴甲基)-2-丁基己酸存在市售价格昂贵且生产厂家偏少,工业化生产路线不成熟等问题;
5.2-(溴甲基)-2-丁基己酸化学结构,式1结构如下:
[0006][0007]
关于2-(溴甲基)-2-丁基己酸的合成路线,主要有以下几类:
[0008]
wo2002008211公开了使用2,2-二正丁基-1,3-丙二醇作为起始原料来制备2-(溴甲基)-2-丁基己酸,反应过程中需要使用不适合工业化生产的钠氢、贵金属催化剂rucl3和使用有毒的四氯化碳作溶剂;该专利报道的方法,起始原料来源困难且反应条件苛刻且需要通过柱层析获得,总收率仅有54%,不适合工业化生产,合成路线如下:
[0009][0010]
文献journal of chemical and pharmaceutical research(2015),7(12),733-740报道使用二丁基丙二酸二乙酯为原料合成2-(溴甲基)-2-丁基己酸,该路线的缺点在于起始原料价格较贵,供应商少,步骤繁琐,收率较低,不适合工业化生产,合成路线如下:
[0011]
[0012]
(a)dibal-h,nabh4;(b)naoh,meoh;(c)hbr,con.h2so4;
[0013]
文献tetrahedron letters(1982),23(31),3151-4报道了2-丁基-2-(羟基甲基)己腈的合成方法,采用还原剂硼氢化锂和催化量的硼氢化钠进行还原得到2-丁基-2-(羟基甲基)己腈。
[0014][0015]
文献journal of the iranian chemical society(2017),14(5),1059-1067报道了通过氰乙酸乙酯,乙醇钠,氯丁烷为原料合成2-丁基-2-氰基己酸乙酯。
[0016][0017]
2-(溴甲基)-2-丁基己酸的合成方法文献报道很少且反应繁琐,收率低,不适合工业化生产。因此,目前需要寻找一种新的2-(溴甲基)-2-丁基己酸工业化合成方法。
技术实现要素:
[0018]
本发明的主要在于提供一种新的2-(溴甲基)-2-丁基己酸的合成方法。
[0019]
2-(溴甲基)-2-丁基己酸
[0020][0021]
经鉴定式(1)化合物的物化特性如下:
[0022]
分子式:c
11h21
bro2,分子量:265.187,沸点:324.3
±
25.0℃ at 760mmhg,白色至类白色固体,无味。
[0023]
本发明提供了一种化合物,其具有如下结构式:
[0024][0025]
本发明提供了式(1)化合物的合成路线如下,
[0026]
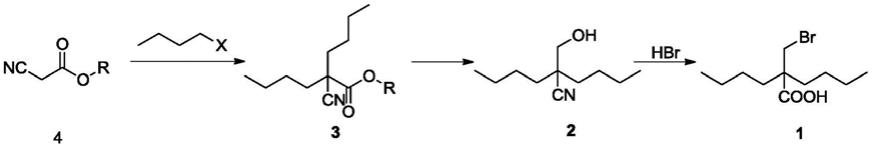
本发明提供一种2-(溴甲基)-2-丁基己酸的合成方法,反应式如下:
[0027]
方法一:
[0028][0029]
方法二:
[0030][0031]
其中r选自甲基或乙基,x选自cl、br或i;
[0032]
所述方法二化合物4合成化合物2为“一锅法”;
[0033]
所述方法一或方法二包含以下步骤:
[0034]
1)化合物4与卤代丁烷在醇钠作用下反应得到化合物3;
[0035]
2)化合物3经还原剂还原得到化合物2;
[0036]
3)化合物2在氢溴酸作用下合成得到化合物1。
[0037]
本发明还提供方法一或方法二优选技术方案,步骤1)中卤代丁烷选自氯代丁烷、溴代丁烷或碘代丁烷;
[0038]
本发明还提供方法一或方法二优选技术方案,步骤1)中醇钠选自甲醇钠、乙醇钠或叔丁醇钠;
[0039]
本发明还提供方法一或方法二优选技术方案,步骤1)在醇溶剂中反应,醇溶剂选自甲醇、乙醇、异丙醇;
[0040]
本发明还提供方法一或方法二优选技术方案,步骤1)在15%-25%质量浓度的乙醇钠乙醇溶液中反应;
[0041]
本发明还提供方法一或方法二优选技术方案,步骤1)中化合物4:卤代丁烷(例如溴代丁烷):醇钠(例如乙醇钠)的摩尔质量比选自1:(2-5):(2-4);优选1:(2.5-3.5):(2.1-3)。
[0042]
本发明还提供方法一优选技术方案,步骤1)选自在20℃至回流温度下反应;
[0043]
本发明还提供方法一优选技术方案,步骤2)反应时间选自3-24小时,优选8-12小时;
[0044]
本发明还提供方法二“一锅法”优选技术方案,步骤1)和步骤2)“一锅法”反应时间选自1-24小时,优选6-12小时,更优选6-8小时;
[0045]
本发明还提供方法一或方法二优选技术方案,步骤2)所述的还原剂选自硼氢化钠、硼氢化锂、硼氢化钙、硼氢化钾、二异丁基铝氢、四氢铝锂、三氟化硼乙醚、硼烷-四氢呋喃、硼烷二甲硫醚一种或其任意组合;
[0046]
本发明还提供方法一或方法二优选技术方案,步骤2)选自在甲醇、乙醇,乙腈,四氢呋喃,dmf,乙醚及各种醚类等;
[0047]
本发明还提供方法一或方法二优选技术方案,步骤2)中还原剂与化合物4摩尔投料比为(1.5-3):1,投料比优选(1.8-2):1;
[0048]
本发明还提供方法一或方法二优选技术方案,步骤2)反应温度选自不高于60℃,优选40-60℃,更优选50-60℃;
[0049]
本发明提供的一种方法二合成一种2-(溴甲基)-2-丁基己酸“一锅法”技术方案,包含以下步骤:化合物4与卤代丁烷在醇钠作用下反应得到化合物3,化合物3不经处理“一锅法”经过还原剂还原得到化合物2,经过在氢溴酸作用下合成得到化合物1。
[0050]
与现有技术相比,本发明采用一锅法合成2-丁基-2-(羟基甲基)己腈后经过简单的溴代/水解反应合成2-(溴甲基)-2-丁基己酸,反应收率高,且只需简单重结晶,能得到纯度好的产品,是一种更适合大规模工业生产新的合成方法。
[0051]
本发明还提供方法一或方法二优选技术方案,在制备化合物2反应中tlc检测反应完全,降温至10-15℃,将反应液加入到冰水中,搅拌有大量固体析出,过滤,将滤液浓缩,水相用适合溶剂(例如二氯甲烷)萃取1-3次,合并有机相,饱和食盐水洗,干燥,过滤浓缩后得2-丁基-2-(羟基甲基)己腈。
[0052]
本发明还提供方法一或方法二优选技术方案,步骤3)氢溴酸选自氢溴酸水溶液,优选30-40%氢溴酸水溶液;
[0053]
本发明还提供方法一或方法二优选技术方案,步骤3)氢溴酸反应中,可选择性加入一定量浓硫酸;优选在40%hbr溶液与浓硫酸混合体系下反应,进一步优选40%hbr溶液与浓硫酸反应体积比选自2:1-50:1,体积比优选2:1-3:1,2:1-4:1,2:1-5:1,2:1-10:1;3:1-4:1,3:1-5:1,3:1-10:1,3:1-50:1;4:1-5:1,4:1-10:1,4:1-50:1;5:1-10:1,5:1-50:1;10:1-50:1,最优体积比2:1,3:1,4:1,5:1,10:1或50:1。
[0054]
本发明还提供方法一或方法二优选技术方案,步骤3)在100-150℃进行反应,优选在130-135℃反应;
[0055]
本发明还提供方法一或方法二优选技术方案,步骤3)反应时间为1-24h,优选1-12小时。
[0056]
本发明提供了式(1)化合物的合成路线,用于在制备胆汁酸转运抑制剂依洛昔巴特elobixibat药物上应用。
[0057]
本发明有益的技术效果:
[0058]
与现有技术相比,本发明提供了一种2-(溴甲基)-2-丁基己酸中间体的合成方法或一种2-(溴甲基)-2-丁基己酸中间体的“一锅法”合成方法,本发明原料易得,价格低廉,本发明人通过巧妙地路线设计,本发明合成的式(1)所示2-(溴甲基)-2-丁基己酸中间体,经过取代,还原,溴代分步获得产品。缩短了反应步骤,该合成方法收率高,能得到高质量2-(溴甲基)-2-丁基己酸,未使用钠氢、贵金属催化剂和其它有毒有害溶剂;本发明合成方法也无需柱层析,适合工业化生产。
[0059]
本发明优选的技术方案提供了“一锅法”合成2-丁基-2-(羟基甲基)己腈化合物2中间体,缩短了生产时间,减少后处理纯化操作,同时原料易得成本低,反应条件温和,后处理简单,可以减少柱层析对产品的损失,减轻精制压力,降低成本,可以显著提高产率,总收率达到90%,纯度98%以上,更适宜工业化大规模生产。
具体实施方式
[0060]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并
不能因此而理解为对本发明专利范围的限制。
[0061]
实施例1:式(2)化合物的分步合成制备
[0062]
式(2)化合物分步合成路线,反应式如下:
[0063][0064]
式(3)化合物的合成过程如下:
[0065]
起始原料:化合物4(氰乙酸乙酯)
[0066][0067]
将23.7g na块切成小块在室温下缓慢地加入到8000ml乙醇中搅拌至na块消失,继续搅拌2h,得乙醇钠溶液。在氮气保护下将制得的乙醇钠溶液室温下缓慢滴加到化合物4氰乙酸乙酯500g和溴丁烷2.0kg的混合体系中,滴加完毕后加热回流到70-80℃反应16小时,取样tlc检测反应完全,将乙醇旋干后向旋干物中加入冰水5000ml搅拌,用mtbe(5000mlx3)萃取,合并有机相后用饱和食盐水(1500mlx2)洗涤,无水硫酸钠干燥后浓缩得油状物化合物3粗品,可直接进行下步反应。
[0068][0069]
取上述油状物85g化合物3溶解在300ml乙醇中,在室温下分7次加入28.54g nabh4,每次间隔15min,加完后在40-60℃下搅拌12小时,取样tlc检测反应完全。加水淬灭旋干有机溶剂后用二氯甲烷(300mlx3)提取,合并有机相后用饱和食盐水(150mlx2)洗涤,无水硫酸钠干燥后浓缩得油状物化合物2粗品,得59.75g油状物化合物2,收率87.5%,纯度98%。
[0070]
实施例2:式(1)化合物“一锅法”合成路线,反应式如下:
[0071][0072]
式(2)化合物的合成过程如下:
[0073]
起始原料:化合物4(氰乙酸乙酯)
[0074][0075]
于50l的三口瓶中加入2.0kg氰乙酸乙酯,6.056kg溴丁烷,升温至45-50℃,开始滴加etona/etoh(~17%,17.68l)溶液,放热明显,控制滴加速度保持反应体系温度小于60℃,滴加完毕后保温50-60℃反应3h,tlc检测反应完全,降温至35-40℃,加入meoh(5~7l),升温加入1.26kg nabh4保持反应温度不高于60℃,加完后保温50℃反应3h,tlc检测反应完全,降温至10-15℃,将反应液加入到20l的冰水中,搅拌2小时有大量固体析出,过滤,将滤液浓缩,水相用二氯甲烷萃取三次,合并有机相,饱和食盐水洗,干燥,过滤浓缩后得2-丁基-2-(羟基甲基)己腈油状物2.8kg,收率92%。
[0076]
化合物1(2-(溴甲基)-2-丁基己酸)的合成
[0077][0078]
于10l的三口瓶中加入1050g的2-丁基-2-(羟基甲基)己腈和hbr(7.0l,40%,水溶液,搅拌后缓慢加入浓硫酸(1.4l),滴加完毕后在130-135℃反应12小时,tlc检测反应完全,降温至10-35℃,加入5l的dcm和200g活性炭,搅拌0.5小时后,过滤,加入300ml的饱和na2co3调节ph为3-5,有机相用水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩得1.5kg红棕色油状物,石油醚重结晶得化合物1 2-(溴甲基)-2-丁基己酸类白色固体,收率76.8%,纯度99.5%。核磁解析数据1h nmr(cdcl3)6 0.89(t,j=7.0hz,6h),1.11-1.24(m,4h),1.25-1.34(m,4h),1.65-1.69(m,4h),3.56(s,2h).
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。