1.本发明涉及药物合成技术领域,具体涉及一种辛酸拉尼米韦的制备方法。
背景技术:
2.辛酸拉尼米韦(laninamivir)是biota pharmaceuticals和daiichi sankyo公司研发的一种神经氨酸酶抑制剂,可用于治疗对奥司他韦(达菲)具有抗药性的流感病毒感染。于2010年获得批准以inavir名称在日本上市。
3.辛酸拉尼米韦化学结构式如下:
[0004][0005]
辛酸拉尼米韦对h1n1、h5n1、n9和b型流感病毒以及对达菲耐药病毒均有较好的效果,截至目前,辛酸拉尼米韦在国内未上市,也没有注册申报厂家。为引导仿制药研发生产,提高公众用药可及性,国家食品药品监督管理总局药品审评中心组织对国内化合物专利权到期、终止、无效且尚无仿制申请的国外已上市药品进行筛选,其中辛酸拉尼米韦初步筛选并纳入《首批专利权到期、终止、无效尚且无仿制申请的药品清单》。
[0006]
目前,辛酸拉尼米韦的制备方法归结为如下三种方法:
[0007]
方法一,专利cn101679339a公开了以唾液酸为起始物料经11步反应合成辛酸拉尼米韦,合成路线如下所示:
[0008]
[0009][0010]
r1=c1~c
19
,r2=c1~c4,r3,r6,r7=c1~c6,r4,r5=四亚甲基、亚戊基或氧代基;
[0011]
该路线以商业化供应的唾液酸为起始物料,经11步反应合成辛酸拉尼米韦,化合物4-6生产过程中反应步骤繁琐且需要柱层析,若要工业化仍需要进一步的优化。
[0012]
方法二,专利cn103435582a公开了以扎那米韦为起始物料经5步反应合成辛酸拉尼米韦,合成路线如下所示:
[0013][0014]
该方法虽然步骤少,但是以扎纳米韦为起始原料,原料的价格过于昂贵,难以满足仿制药的需求;
[0015]
方法三,马大伟课题组以d-异抗坏血酸为起始物料,经手性催化关环,再经一系列氧化、还原、保护、脱保护等反应合成辛酸拉尼米韦,合成路线如下所示:
[0016][0017]
该合成路线比较新颖,单元反应收率适中,但起始物料及催化剂配体市场供应较少,价格昂贵,不适合放大生产,同时中间体s-3和中间体s-8手性纯度不高,导致后续异构体杂质较多,最终产品质量难以控制。
[0018]
综上,上述三条路线均不适合工业化生产,开发一条适合工业化生产辛酸拉尼米韦的路线将具有非常好的应用前景。
技术实现要素:
[0019]
本发明的目的在于提供一种辛酸拉尼米韦的制备方法,该方法工艺简单且收率高,适合工业化生产。
[0020]
为实现上述目的,本发明的技术方案是:
[0021]
一种辛酸拉尼米韦的制备方法,采用包括如下的合成路线:
[0022][0023]
其中,r1为苄基、取代苄基或烯丙基;
[0024]
化合物13和化合物14的结构式如下所示:
[0025][0026]
上述取代苄基为苄基上的苯环被氯、溴、碘、硝基、烷氧基、烷基、芳香基等基团单取代或多取代。
[0027]
上述式(4)所示化合物采用包括如下的合成路线得到:
[0028][0029]
其中,x为cl-、br-、i-、meso
4-、tfo-。
[0030]
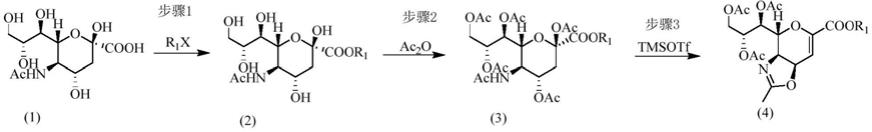
步骤1,在碱存在下,式(1)所示的化合物与r1x反应生成式(2)表示的化合物。所述碱没有特别的限制,可以与羧酸中和即可,可以是有机碱或无机碱。优选的,所述有机碱诸如三乙胺、二异丙基乙胺、n,n-二甲基-4-氨基吡啶或吡啶;无机碱诸如碳酸铯、碳酸钾、碳酸钠、氢氧化钠、氢氧化钾。优选无机碱,且进一步优选碳酸铯或碳酸钾。
[0031]
步骤2,在有机碱存在下,式(2)表示的化合物与乙酸酐反应,生成式(3)表示的化合物。关于有机碱没有限制,可以与羧酸中和即可。
[0032]
优选的,所述有机碱为三乙胺、二异丙基乙胺、n,n-二甲基-4-氨基吡啶、吡啶中的任意一种或几种。
[0033]
步骤2的反应温度为-30℃~100℃。优选为0~35℃。
[0034]
步骤2的反应时间是30min~24h。优选为10~15h。
[0035]
步骤3,式(3)表示的化合物在三氟甲磺酸三甲硅酯催化下,产生式(4)表示的化合物。
[0036]
步骤3的反应温度0~70℃。优选为30℃~60℃。
[0037]
步骤3的反应时间是1.0h~24h。优选为1.0~3.0h。
[0038]
步骤3后处理滴加三乙胺淬灭反应。
[0039]
步骤4为将式(4)所示化合物与有机溶剂、r1oh混合搅拌,之后在氮气保护下,加入nah搅拌30~60min,然后加入(r1o)2co,于0~80℃搅拌反应30min~24h,之后分离,得式(5)所示化合物。式(5)化合物粗品经甲苯或甲醇结晶,可得式(5)表示化合物的晶体。
[0040]
优选的,上述步骤4的反应温度为10~60℃。进一步优选的,所述反应温度为50~55℃。
[0041]
优选的,上述步骤4的反应时间为30min~5h。
[0042]
步骤4中所述有机溶剂为甲苯、n,n-二甲基甲酰胺、苯甲醇、正庚烷、甲基叔丁基醚、四氢呋喃、1,4-二氧六环中的任意一种或几种。
[0043]
步骤4中nah的质量浓度为60%。
[0044]
步骤4中,式(4)所示化合物、r1oh、nah、(r1o)2co的质量比为8-12:15-30:0.02-0.1:12-36。优选地,式(4)所示化合物、r1oh、nah、(r1o)2co的质量比为9.8:20:0.04:24.22。
[0045]
步骤5是在碱存在下,式(5)表示的化合物与硫酸二甲酯或碘甲烷反应,产生式(6)表示的化合物。
[0046]
步骤5反应温度为0-70℃,优选0-30℃。
[0047]
步骤5反应时间为30min~24h,优选1.0~15h。
[0048]
步骤6是在异丙醇钛催化下使式(6)表示的化合物与叠氮三甲基硅烷反应,以产生式(7)表示的化合物。
[0049]
步骤6反应温度为0~120℃,优选为0~40℃。
[0050]
步骤6反应时间为3.0~48h,优选3.0~24h。
[0051]
步骤7是式(7)表示的化合物与三苯基膦反应以产生式(8)表示的化合物。
[0052]
步骤7反应温度为-10~80℃,优选0~60℃。
[0053]
步骤7反应时间为10分钟~48h,优选10min~5h。
[0054]
步骤8是式(8)表示的化合物与碱反应以产生式(9)表示的化合物。
[0055]
关于碱,优选无机碱,且更优选氢氧化锂、氢氧化钠、氢氧化钾。
[0056]
步骤8反应温度优选-10~100℃,进一步优选0~50℃。
[0057]
步骤8反应时间优选10分钟~48h,进一步优选0.5h~6h。
[0058]
步骤9是式(9)表示的化合物与式(10)表示的化合物反应以产生式(11)表示的化合物。
[0059]
步骤9反应溶剂优选纯化水。
[0060]
步骤9反应温度是0-100℃,且优选5~50℃。
[0061]
步骤9反应时间是24h~72h,且优选24~60h。
[0062]
步骤10是式(11)表示的化合物在三氟乙酸存在下反应,以产生式(12)表示的化合物。
[0063]
步骤10反应温度是-20~100℃,且优选0~45℃。
[0064]
步骤10反应时间是0.5h~24h,且优选0.5~8h。
[0065]
步骤10需要调ph至6~8。
[0066]
步骤11是在酸存在下使式(12)表示的化合物与化合物13或化合物14反应,之后再与水反应,以产生由式(ⅰ)表示的化合物或其药理学可接受的盐。
[0067]
步骤11中的酸优选无机酸,且更优选盐酸。
[0068]
步骤11反应温度优选-10~70℃,且优选0~50℃。
[0069]
步骤11反应时间优选10分钟至24小时,且更优选10分钟至5小时。
[0070]
化合物13和化合物14可以通过现有技术中的制备方法制得;也可以通过如下路线制得:
[0071][0072]
优选的,r1为苄基。
[0073]
本发明的有益效果:
[0074]
本发明辛酸拉尼米韦的制备方法,创新性地采用一锅法由化合物(4)合成辛酸拉尼米韦中间体化合物(5),收率高达87%以上,而采用cn101679339a中的两步反应的产率仅为76.9%,本发明反应温和快速,并且产物无需柱层析,简单后处理即可进行下一步反应,节省能耗,减少污染,操作简单,成本低,适合工业化生产。本发明最终制得的辛酸拉尼米韦的收率在85%以上。
附图说明
[0075]
图1为化合物(2)的氢谱图;
[0076]
图2为化合物(3)的氢谱图;
[0077]
图3为化合物(4)的氢谱图;
[0078]
图4为化合物(5)的氢谱图;
[0079]
图5为化合物(6)的氢谱图;
[0080]
图6为化合物(12)的氢谱图;
[0081]
图7为化合物(13)的氢谱图。
具体实施方式
[0082]
本发明辛酸拉尼米韦的制备方法,采用包括如下的合成路线:
[0083][0084]
其中,r1为苄基、取代苄基或烯丙基;
[0085]
化合物13和化合物14的结构式如下所示:
[0086][0087]
上述取代苄基,指:苄基上的苯环被氯、溴、碘、硝基、烷氧基、烷基、芳香基等基团单取代或多取代。
[0088]
上述式(4)所示化合物采用包括如下的合成路线得到:
[0089][0090]
其中,x为cl-、br-、i-、meso
4-、tfo-。
[0091]
步骤1,在碱存在下,式(1)所示的化合物与r1x反应生成式(2)表示的化合物。所述碱没有特别的限制,可以与羧酸中和即可,可以是有机碱或无机碱。优选的,所述有机碱诸如三乙胺、二异丙基乙胺、n,n-二甲基-4-氨基吡啶或吡啶;无机碱诸如碳酸铯、碳酸钾、碳酸钠、氢氧化钠、氢氧化钾。优选无机碱,且进一步优选碳酸铯或碳酸钾。
[0092]
步骤2,在有机碱存在下,式(2)表示的化合物与乙酸酐反应,生成式(3)表示的化合物。关于有机碱没有限制,可以与羧酸中和即可。
[0093]
优选的,所述有机碱为三乙胺、二异丙基乙胺、n,n-二甲基-4-氨基吡啶、吡啶中的任意一种或几种。
[0094]
步骤2的反应温度为-30℃~100℃。优选为0~35℃。
[0095]
步骤2的反应时间是30min~24h。优选为10~15h。
[0096]
步骤3,式(3)表示的化合物在三氟甲磺酸三甲硅酯催化下,产生式(4)表示的化合物。
[0097]
步骤3的反应温度0~70℃。优选为30℃~60℃。
[0098]
步骤3的反应时间是1.0h~24h。优选为1.0~3.0h。
[0099]
步骤3后处理滴加三乙胺淬灭反应。
[0100]
步骤4为将式(4)所示化合物与有机溶剂、r1oh混合搅拌,之后在氮气保护下,加入nah搅拌30~60min,然后加入(r1o)2co,于0~80℃搅拌反应30min~24h,之后分离,得式(5)所示化合物。式(5)化合物粗品经甲苯或甲醇结晶,可得式(5)表示化合物的晶体。
[0101]
优选的,上述步骤4的反应温度为10~60℃。进一步优选的,所述反应温度为50~55℃。
[0102]
优选的,上述步骤4的反应时间为30min~5h。
[0103]
步骤4中所述有机溶剂为甲苯、n,n-二甲基甲酰胺、苯甲醇、正庚烷、甲基叔丁基醚、四氢呋喃、1,4-二氧六环中的任意一种或几种。
[0104]
步骤4中nah的质量浓度为60%。
[0105]
步骤4中,式(4)所示化合物、r1oh、nah、(r1o)2co的质量比为8-12:15-30:0.02-0.1:12-36。优选地,式(4)所示化合物、r1oh、nah、(r1o)2co的质量比为9.8:20:0.04:24.22。
[0106]
步骤5是在碱存在下,式(5)表示的化合物与硫酸二甲酯或碘甲烷反应,产生式(6)表示的化合物。
[0107]
步骤5反应温度为0-70℃,优选0-30℃。
[0108]
步骤5反应时间为30min~24h,优选1.0~15h。
[0109]
步骤6是在异丙醇钛催化下使式(6)表示的化合物与叠氮三甲基硅烷反应,以产生式(7)表示的化合物。
[0110]
步骤6反应温度为0~120℃,优选为0~40℃。
[0111]
步骤6反应时间为3.0~48h,优选3.0~24h。
[0112]
步骤7是式(7)表示的化合物与三苯基膦反应以产生式(8)表示的化合物。
[0113]
步骤7反应温度为-10~80℃,优选0~60℃。
[0114]
步骤7反应时间为10分钟~48h,优选10min~5h。
[0115]
步骤8是式(8)表示的化合物与碱反应以产生式(9)表示的化合物。
[0116]
关于碱,优选无机碱,且更优选氢氧化锂、氢氧化钠、氢氧化钾。
[0117]
步骤8反应温度优选-10~100℃,进一步优选0~50℃。
[0118]
步骤8反应时间优选10分钟~48h,进一步优选0.5h~6h。
[0119]
步骤9是式(9)表示的化合物与式(10)表示的化合物反应以产生式(11)表示的化合物。
[0120]
步骤9反应溶剂优选纯化水。
[0121]
步骤9反应温度是0-100℃,且优选5~50℃。
[0122]
步骤9反应时间是24h~72h,且优选24~60h。
[0123]
步骤10是式(11)表示的化合物在三氟乙酸存在下反应,以产生式(12)表示的化合物。
[0124]
步骤10反应温度是-20~100℃,且优选0~45℃。
[0125]
步骤10反应时间是0.5h~24h,且优选0.5~8h。
[0126]
步骤11是在酸存在下使式(12)表示的化合物与化合物13或化合物14反应,之后再
与水反应,以产生由式(ⅰ)表示的化合物或其药理学可接受的盐。
[0127]
步骤11中的酸优选无机酸,且更优选盐酸。
[0128]
步骤11反应温度优选-10~70℃,且优选0~50℃。
[0129]
步骤11反应时间优选10分钟至24小时,且更优选10分钟至5小时。
[0130]
化合物13和化合物14可以通过现有技术中的制备方法制得;也可以通过如下路线制得:
[0131][0132]
优选的,r1为苄基。
[0133]
实施例1
[0134]
化合物(2)的制备包括以下步骤:
[0135]
常温,于500ml反应瓶中依次加入n-乙酰神经氨酸(化合物1)30.93g、cs2co
3 16.30g、125mldmf、brbn 26.70g,磁力搅拌反应24h,待反应结束后,过滤,母液浓缩至干,加入300ml异丙醇,升温至70℃,热滤出去不溶物,滤液常温搅拌3-4h,过滤,滤饼用异丙醇淋洗,滤饼减压干燥恒重得化合物(2)白色粉末18.50g,收率46.25%。化合物(2)的氢谱图如图1所示。
[0136]
在其它实施例中,化合物(2)的制备过程中,加入300ml异丙醇后,还可以升温至80℃或75℃,然后再热滤出去不溶物。
[0137]
实施例2
[0138]
化合物(3)的制备包括以下步骤:
[0139]
常温,于250ml反应瓶中,依次加入吡啶约70ml、化合物(2)18.50g,加毕控温20~25℃范围内,滴加醋酸酐37.50g,之后,加入4-二甲氨基吡啶(dmap)约0.2g,维持20~25℃,搅拌反应过夜,反应液倒入350ml纯化水与1850ml乙酸乙酯混合液中,加毕,静置分层,有机层依次用200ml*2 5.0%hcl水溶液萃洗、200ml*2饱和碳酸氢钠水溶液萃洗,之后有机层无水硫酸钠干燥,过滤,淋洗,减压浓缩至干得无色油状物30.02g,收率106.4%。经柱层析(正庚烷~正庚烷/乙酸乙酯=1/2)纯化,收集目标组分(展开剂:乙酸乙酯),减压浓缩干,得化合物(3)白色粉末14.50g。化合物(3)的氢谱图如图2所示。
[0140]
实施例3
[0141]
化合物(4)的制备包括以下步骤:
[0142]
常温,n2保护下,于1000ml反应瓶中,依次加入化合物(3)13.20g、乙酸乙酯265ml,搅拌溶解澄清,控温15~25℃,滴加tmsotf 14.46g,滴加结束,控温50~55℃范围内,搅拌反应2h,然后控温0~5℃,滴加三乙胺11.0g,之后加入冰水100ml,搅拌10min,静置分层,有机层再用100ml 50ml纯化水萃洗两次,有机层无水硫酸钠干燥,过滤,淋洗,减压浓缩至干,得无色油状物。柱层析纯化(正庚烷~正庚烷/乙酸乙酯=1/1~乙酸乙酯)收集目标组分,
减压浓缩至干得化合物(4)无色油状物9.14g。化合物(4)的氢谱图如图3所示。
[0143]
实施例4
[0144]
化合物(5)的制备包括以下步骤:
[0145]
常温,于反应瓶中依次加入化合物(4)9.80g、甲苯40ml、苯甲醇(即r1取苄基)20.0ml,加毕,室温搅拌,氮气保护,之后,加入60%nah 0.04g,搅拌30min,加入碳酸二苄酯(即r1取苄基)24.22g,于50℃搅拌反应1h,加入0.032mol冰乙酸淬灭反应,湿法上柱,二氯甲烷冲洗小极性杂质,二氯甲烷/甲醇=40/1,洗刷目标组分,减压浓缩干得化合物(5)类白色固体,6.85g,收率87.96%。该收率远远高于cn101679339a中两步法得化合物(5)的收率(76.9%)。化合物(5)的氢谱图如图4所示。
[0146]
在其它化合物(5)的制备实施例中,加入碳酸二苄酯后于55℃搅拌反应2h。在其他的实施例中,搅拌反应温度还可以为10℃、52℃、60℃或80℃。搅拌反应时间可以在30min~24h内调整。改变上述反应温度或时间,化合物(5)的收率与实施例4基本相同。
[0147]
在其他实施例中,r1还可以为被氯、溴、碘、硝基、烷氧基、烷基或芳香基取代的苄基;甚至r1还可以为烯丙基。
[0148]
在其他的化合物(5)的制备实施例中,式(4)所示化合物、r1oh、nah、(r1o)2co的质量比为8:30:0.02:36。
[0149]
在其他的化合物(5)的制备实施例中,式(4)所示化合物、r1oh、nah、(r1o)2co的质量比为12:15:0.1:12。
[0150]
在其它化合物(5)的制备实施例中,甲苯溶剂还可以被替换为n,n-二甲基甲酰胺、苯甲醇、正庚烷、甲基叔丁基醚、四氢呋喃、1,4-二氧六环中的任意一种或几种。
[0151]
实施例5
[0152]
化合物(6)的制备包括以下步骤:
[0153]
n2保护下,于250ml反应瓶中,依次加入化合物(5)6.85g、thf 28ml、dmf 7ml,加毕,搅拌至溶解澄清,控温0-5℃范围内,分批次加入60%nah 0.91g,加毕维持温度0-5℃,搅拌30min,加入硫酸二甲酯2.88g,维持温度搅拌反应过夜,之后加入甲苯100ml、冰乙酸1.06g淬灭反应,反应液用40ml*2 5.0%nahco3水溶液萃洗2次,水层用70ml*2甲苯萃洗2次,合并所有甲苯层无水硫酸钠干燥,过滤,淋洗,浓缩至近干,柱层析纯化(二氯甲烷
→
二氯甲烷/甲醇20/1),得化合物(6)无色油状物8.0g。化合物(6)的氢谱图如图5所示。
[0154]
实施例6
[0155]
化合物(7)的制备包括以下步骤:
[0156]
n2保护下,室温,于100ml反应瓶中,依次加入化合物(6)7.10g、甲苯21ml、叔丁醇7ml,加毕,搅拌至溶解澄清,加入tmsn
3 4.10g、异丙醇钛1.55g,维持20~25℃搅拌反应24h,之后过滤,滤液抽干,10ml甲醇淋洗,油泵带干滤饼,得化合物(7)米白色固体5.76g,收率:72.6%。
[0157]
实施例7
[0158]
化合物(8)的制备包括以下步骤:
[0159]
常温,于100ml反应瓶,依次加入化合物(7)5.00g、四氢呋喃20ml,搅拌至溶清,加入三苯基膦3.24g,加毕升温至40~50℃,搅拌反应10min,tlc(dcm/me=10/1),原料消失,降至室温,得化合物(8)反应液。
[0160]
实施例8
[0161]
化合物(9)的制备包括以下步骤:
[0162]
于实施例7得到的化合物(8)的反应液中,加入纯化水12.5g、25%naoh水溶液5.40g,控温40~45℃,搅拌反应2h,tlc(dcm/me=10/1),原料消失,降至室温,静置分液,水层加入6mlthf萃洗一次,水层滴加约4ml浓盐酸,调ph=2~3,再用25%naoh水溶液调ph=9~10,得化合物(9)反应液。
[0163]
实施例9
[0164]
化合物(11)的制备包括以下步骤:
[0165]
室温,于实施例8的化合物(9)反应液中,加入甲醇30ml、n,n
’-
双(叔丁氧羰基)-1h-吡唑-1-甲脒3.82g,维持20~25℃搅拌反应48h,调ph=8.5~8.8,反应液浓缩至尽干,加入100ml乙酸乙酯减压浓缩带除甲醇,浓缩液用50ml*3乙酸乙酯萃洗3次,水层用约1.3g浓盐酸调ph=2~3,水层用50ml*3乙酸乙酯萃取,合并有机层浓缩干,得白色固体5.20g,收率85.1%。
[0166]
实施例10
[0167]
化合物(12)的制备包括以下步骤:
[0168]
将实施例9中化合物(11)加入50ml二氯甲烷,加入5g三氟乙酸室温反应3h,减压浓缩至干,加入25ml纯化水,用10%氢氧化钠水溶液调至ph=6~8,加入50ml甲醇,常温搅拌析晶,过滤,滤饼甲醇淋洗,40℃减压干燥得化合物(12)白色粉末2.74g,收率70.62%。
[0169]
实施例11
[0170]
常温,n2保护下,与100ml反应瓶中,依次加入化合物(12)2.00g、甲醇10ml、3.54g化合物13,最后加入10.5ml hcl甲醇溶液(1n氯化氢甲醇溶液),加毕,体系澄清,维持20-25℃,搅拌反应1h,减压浓缩近干,加入20ml纯化水,用20ml*3乙酸乙酯萃洗三次,水层用饱和碳酸钠调ph=7,搅拌1h,之后,用饱和碳酸钠调ph=8.5~9.0,搅拌3h,用6n盐酸调ph=5~6,搅拌30min,过滤,滤饼纯化水淋洗,滤饼35℃,减压干燥至恒重,得化合物(i)白色粉末2.34g,收率85.6%,hplc 99.84%。
[0171]
本实施例中的化合物13可以替换为化合物14,其它条件不变。
[0172]
对比例1
[0173]
该对比例与实施例4的区别仅在于,将碳酸二苄酯替换为羰基二咪唑。产物杂质较多,需要柱层析,最终得到的化合物(5)的收率仅为20%。
[0174]
对比例2
[0175]
该对比例与实施例4的区别仅在于,将碳酸二苄酯替换为氯甲酸苄酯。产物杂质较多,需要柱层析,最终得到的化合物(5)的收率仅为25%。
[0176]
对本领域的技术人员来说,可根据以上描述的技术方案以及构思,做出其它各种相应的改变以及形变,而所有的这些改变以及形变都应该属于本发明权利要求的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

