1.本发明涉及化学合成领域,具体而言涉及一种合成芳香族化合物的方法
背景技术:
2.芳烃是重要的化工产品,可直接用作溶剂,也是合成其他重要化工原料的中间体,如乙苯是生产苯乙烯的原料,可用于制药和有机合成领域。正丙苯可用于纺织染料、印刷、醋酸纤维溶剂及合成聚丙烯成核剂的中间体。芳香族羧酸在有机合成,香料及医药领域有重要的应用。如苯丙酸是香料的定香剂,也可用作医药中间体,苯丁酸则在合成染料,医药领域有重要应用。
3.friedel-crafts酰基化反应在有机合成及药物合成中是一类非常重要的反应,传统的friedel-crafts酰基化反应制备通常使用lewis酸催化酰氯与芳香类化合物,这也导致过程中需要使用大量的均相催化剂,以酰氯为原料后处理过程会产生大量hcl气体,导致环境污染。并且反应会产生大量的铝盐废液。目前已有以杂多酸或非均相催化剂进行羧酸或酸酐与芳香族化合物friedel-crafts酰基化反应的相关报道,在多个报道中均发现在非均相催化剂下积碳会迅速生成会导致催化剂活性下降。积碳主要是羧酸或酸酐与芳香族化合物进行friedel-crafts酰基化反应生成的酮类化合物在催化剂的酸性位上形成的(chemical communications.,2003,530-531,journal of catalysis,1999,187,209-218,catalysis letters,2008,126,188-192)。若要由上述芳香族酮类产物得到芳烃或芳香族羧酸则需要进行还原,如苯丁酸的合成可采用苯与酸酐经过傅克酰基化之后再经过定量还原试剂进行还原得到。cn201810564276.0公开了以水合肼为还原剂还原4-氧代-4-苯基丁酸制备4-苯基丁酸的过程。但上述过程中均为分步进行且还原步骤需采用定量还原试剂如硼氢化钠、硼氢化钾、水合肼等进行还原,会产生大量的废水,造成环境污染。
技术实现要素:
4.本发明的技术目的是提供一种制备芳香族化合物的方法。
5.一方面,本发明提供一种制备芳香族化合物的方法,所述方法包括以下步骤:
6.1)在固定床反应器中加入成型的催化剂,在还原气气氛下升温至催化剂活化温度下还原4-12h后调整至反应温度;
7.2)将反应原料a和反应原料b分别经过预热后泵入固定床反应器中进行反应;以及
8.3)反应产物经过冷凝和气液分离后精馏得到最终产品,
9.其中,在步骤1)中,所述催化剂包括金属活性组分和催化剂载体,所述金属活性组分包括pd、pt、ru、rh、ir、ni、cu、co中的至少一种;所述催化剂载体包括γ-氧化铝、二氧化硅、五氧化二铌、三氧化钨、二氧化锆、分子筛中的至少一种,优选地,所述催化剂载体为zsm-5或γ-al2o3;并且基于催化剂载体的重量,金属活性组分的负载量为1-30wt%;
10.在步骤2)中,所述反应原料a选自苯和取代苯,所述反应原料b选自羧酸和酸酐,
11.所述取代苯指苯环的1个或2个氢原子被取代基取代的苯,并且所述取代基选自
c1-c8烷基、羟基以及c1-c8烷氧基,优选地,选自c1-c6烷基、羟基以及c1-c6烷氧基,
12.所述羧酸包括具有2-12个,优选3-10个,更优选4-8个碳原子的取代或未取代的单羧酸及具有3-12个,优选4-10个,更优选5-8个碳原子的取代或未取代的二羧酸,
13.所述酸酐包括具有2-12个,优选3-10个,更优选4-8个碳原子的取代或未取代的单羧酸酸酐及具有3-12个,优选4-10个,更优选5-8个碳原子的取代或未取代的二羧酸酸酐,
14.其中,所述羧酸、二羧酸和酸酐、二酸酐中的取代基可包括c1-c6烷基、羟基、氨基、c1-c6烷氧基、c6-c10芳基或含n、p、s杂原子的c5-c10杂芳基,优选地,可包括c1-c3烷基、羟基、氨基、c1-c3烷氧基、c6芳基或含n、p、s杂原子的c5杂芳基,
15.其中,所述芳香族化合物是指芳环上具有取代基的化合物,所述取代基可选自c1-c6烷基、c2-c6烯基、c1-c6烷氧基、-c(=o)c1-c6烷基、氧代基团、羟基中的一种或多种,其中所述c1-c6烷基、c2-c6烯基、c1-c6烷氧基可进一步被选自羧基、羟基中的一种或多种所取代。
16.在具体实施方式中,在步骤1)中,所述催化剂活化温度为200-500℃,优选为350-450℃;所述反应温度为90-400℃,优选为120-220℃;还原过程中使用氢气,且氢气压力为0.1-7mpa,优选为0.1-3mpa。
17.在具体实施方式中,可在溶剂存在或不存在下进行步骤2),在存在溶剂的情况下,所述溶剂可选自水、甲醇、冰醋酸、丙酮、乙醚、硝基苯、二氯乙烷、石油醚、二硫化碳、四氯化碳、乙醇、异丙醇、1,4-二氧六环、四氢呋喃以及乙腈中的一种或多种。
18.在具体实施方式中,在步骤2)中,反应原料a与反应原料b的摩尔比可为1:1-1:20,优选1:1-1:5。
19.在具体实施方式中,在步骤2)中,原料的进料空速为0.01-30h-1
,优选为0.1-1h-1
。
20.在具体实施方式中,在步骤1)中所使用的催化剂通过包括以下步骤的方法制备:
[0021]1’
):催化剂载体预处理:将催化剂载体浸渍在选自硝酸铵或磷酸的溶液中以进行预处理,以及进行真空干燥和焙烧;
[0022]2’
):金属活性组分的负载、冷冻成型及冷冻干燥:将含有金属活性组分的金属前驱体配制成溶液,将上述催化剂载体加入该溶液中,搅拌混合后,加入液氮中冷冻成型,再将成型后的固体置于冷冻干燥机中干燥,研磨成粉末;
[0023]3’
):微波反应:将步骤2’)中得到的粉末置于微波反应器中处理;
[0024]4’
):等离子体处理:将步骤3’)中的所得物平铺加入石英反应釜中,置于两电极之间,用反应气体置换釜内空气,调节电压电流以对样品进行处理,将处理后的样品焙烧即得催化剂。
[0025]
在具体实施方式中,在步骤1’)中,硝酸铵或磷酸水溶液的浓度为0.1-2mol/l;载体与硝酸铵或磷酸水溶液的质量比为1:10-1:100;真空干燥温度为70-150℃,时间为4-15h;焙烧温度为300-450℃,时间为5-12h。
[0026]
在具体实施方式中,在步骤1’)中,所述催化剂载体包括γ-氧化铝、二氧化硅、五氧化二铌、三氧化钨、二氧化锆、分子筛(例如zsm-5、zsm-35)中的至少一种,优选地,所述催化剂载体为zsm-5、五氧化二铌或γ-al2o3中的至少一种。
[0027]
在具体实施方式中,在步骤2’)中,所述金属活性组分包括pd、pt、ru、rh、ir、ni、cu、co中的至少一种,优选的,所述金属活性组分包括pt、pd、ni、ru中的至少一种。
[0028]
在具体实施方式中,在步骤3’)中,微波反应器功率为100-450w,优选300-400w,微波反应时间5-30min,优选10-20min。
[0029]
在具体实施方式中,在步骤4’)中,所述反应气体包括氮气、氩气、氧气或氢气,优选氮气、氩气。
[0030]
在具体实施方式中,在步骤4’)中,等离子体处理的电压为100-200v,电流为1.5-2.5a,时间为10-100min,优选地,等离子体处理的电压为100-150v,电流为2.0-2.5a,时间为40-60min。
[0031]
有益效果
[0032]
本发明的方法制备的催化剂同时具有金属活性位点与酸性位点,可以使friedel-crafts酰基化反应、酮加氢、醇脱水和烯烃加氢在同一催化剂上进行。
[0033]
此外,催化剂的制备过程中使用等离子体处理可以减少反应过程中积碳的形成。催化剂各种活性位点之间的协同作用减少了friedel-crafts酰基化反应生成的酮类产物与催化剂的接触,也减少积碳的生成,增强了催化剂的稳定性。
[0034]
总之,本发明提供的制备芳香族化合物的方法具有以下优点:工艺简单,原料易得,效率高,路线环保,可连续生产,同时可以在更长的时间以高转化率及高选择性实现所述芳香族化合物的高效合成。
附图说明
[0035]
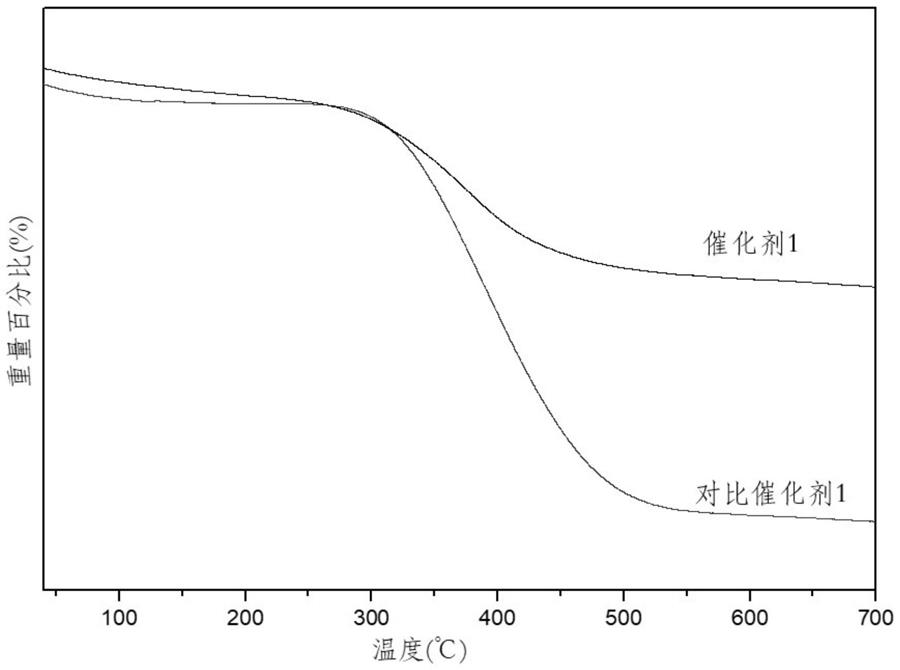
图1为参与反应后的催化剂1与对比催化剂1的热重曲线。
[0036]
图2示出合成实施例1中苯甲醚与醋酸酐反应的稳定性。
[0037]
图3示出对比实施例1中苯甲醚与醋酸酐反应的稳定性。
具体实施方式
[0038]
以下实施例仅是作为本发明的实施方案的例子列举,并不对本发明构成任何限制,本领域技术人员可以理解在不偏离本发明的实质和构思的范围内的修改均落入本发明的保护范围。
[0039]
术语:
[0040]
在本技术中的数值范围,例如“1-6个”,或“2-12个”等可包括其中包含的各具体整数值,比如“1-6”的范围可包括1、2、3、4、5、6,“1-12”的范围可包括1、2、3、4、5、6、7、8、9、10、11和12。
[0041]
在下文中,在制备芳香族化合物的方法中,将制备得到的产物,过0.22μm滤膜,用气相色谱(gc)进行分析检测。气相色谱检测条件:仪器:岛津gc2010plus,色谱柱:hp-5,30m
×
0.25mm
×
0.25um,汽化室温度250℃,fid温度300℃,柱温箱升温程序:60℃保持1min,然后以15℃/min速度升温至280℃保持10min。通过气质联用(gc-ms)和标准物gc保留时间作为对照对产物进行定性分析。用varian 450-gc气相色谱对产物进行定量测定,通过与标准物保留时间和峰面积大小比对进行定量分析。液体产物的收率以(目标产物的摩尔量)/(原料a的摩尔量)
×
100%进行计算,相关计算公式如下:
[0042]
原料a的转化率(%)=(n
原料a1-n
原料a2
)/n
原料a1
×
100%
[0043]
其中n
原料a1
为反应前原料a的摩尔量,n
原料a2
为反应后原料a的摩尔量;产物收率(%)
=(n
产物
/n
原料a
)
×
100%
[0044]
其中n
产物
为产物的摩尔量。
[0045]
产物的选择性(%)=产物收率/原料a的转化率
×
100%
[0046]
催化剂的制备
[0047]
制备实施例1
[0048]
1.将10g zsm-5在0.5mol/l的硝酸铵溶液中浸渍5h(浸渍3次)后,放入真空烘箱中80℃干燥12h,然后在300℃下焙烧6h。
[0049]
2.将上述处理后的zsm-5催化剂置于0.1mol/l的氯铂酸水溶液中搅拌混合6h后过滤,固体加入液氮中冷冻成型,再将成型后的固体催化剂置于冷冻干燥机中干燥12h,研磨成粉。
[0050]
3.将上述粉末置于微波反应器中,在400w功率下反应20min,得到黑色固体粉末。
[0051]
4.将上述黑色固体粉末平铺加入石英反应釜中,置于两电极之间,用氩气置换釜内空气,调节电压100v电流2.5a处理60min。将上述样品在300℃下焙烧3h即得催化剂1,其热重曲线参见图1,热重分析在耐驰sta449f5jupiter上进行,将催化剂在氮气气氛下以10℃/min的速度进行升温至800℃,氮气流速为150ml/min。
[0052]
制备实施例2
[0053]
1.将10g zsm-5在0.5mol/l的硝酸铵溶液中浸渍5h(浸渍3次)后,放入真空烘箱中80℃干燥12h,然后在300℃下焙烧6h。
[0054]
2.将上述处理后的zsm-5催化剂置于0.1mol/l的硝酸钯水溶液中搅拌混合6h后过滤,固体加入液氮中冷冻成型,再将成型后的固体催化剂置于冷冻干燥机中干燥12h,研磨成粉。
[0055]
3.将上述粉末置于微波反应器中,在400w功率下反应20min,得到黑色固体粉末。
[0056]
4.将上述黑色固体粉末平铺加入石英反应釜中,置于两电极之间,用氩气置换釜内空气,调节电压100v电流2.5a处理60min。将上述样品在300℃下焙烧3h即得催化剂2。
[0057]
制备实施例3
[0058]
1.首先将10gγ-al2o3在1mol/l的磷酸水溶液中浸渍5h后,过滤,放入真空烘箱中80℃干燥12h,然后在400℃下焙烧6h。
[0059]
2.将上述γ-al2o3置于0.1mol/l的氯铂酸水溶液中搅拌混合6h后过滤,固体加入液氮中冷冻成型,再将成型后的固体催化剂置于冷冻干燥机中干燥12h,研磨成粉。
[0060]
3.将上述粉末置于微波反应器中,在400w功率下反应20min,得到黑色固体粉末。
[0061]
4.将上述黑色固体粉末平铺加入石英反应釜中,置于两电极之间,用氩气置换釜内空气,调节电压100v电流2.5a处理60min。将上述样品在300℃下焙烧3h即得催化剂3。
[0062]
对比制备实施例1
[0063]
1.将10g zsm-5在0.5mol/l的硝酸铵溶液中浸渍5h(浸渍3次)后,放入真空烘箱中80℃干燥12h,然后在300℃下焙烧6h。
[0064]
2.将上述处理后的zsm-5催化剂置于0.1mol/l的氯铂酸水溶液中搅拌混合6h后过滤,固体加入液氮中冷冻成型,再将成型后的固体催化剂置于冷冻干燥机中干燥12h,研磨成粉。
[0065]
3.将上述粉末置于微波反应器中,在400w功率下反应20min,得到对比催化剂1,其
热重曲线参见图1。
[0066]
对比制备实施例2
[0067]
1.10gγ-al2o3在1mol/l的磷酸水溶液中浸渍5h后,过滤,放入真空烘箱中80℃干燥12h,然后在400℃下焙烧6h。
[0068]
2.将上述处理后的γ-al2o3置于微波反应器中,在400w功率下反应20min,得到固体粉末。
[0069]
3.将上述固体粉末平铺加入石英反应釜中,置于两电极之间,用氩气置换釜内空气,调节电压100v电流2.5a处理60min。将上述样品在300℃下焙烧3h。即得对比催化剂2。
[0070]
合成实施例
[0071]
合成实施例1
[0072]
1.在固定床反应器中加入1g上述制备实施例1中制备的催化剂1,在常压氢气气氛下升温至400℃并保持3h后降至180℃。
[0073]
2.将苯甲醚与醋酸酐(摩尔比1:2)分别用柱塞泵(空速1.0h-1
)通入预热器预热后进入固定床反应器中进行反应。
[0074]
3.反应产物经过冷凝和气液分离。
[0075]
经gc检测,反应20h时,苯甲醚的转化率为87%,产物中4-乙基苯甲醚选择性90%,2-乙基苯甲醚选择性1%,对甲氧基苯乙酮选择性2%,对甲氧基苯-α-甲基苯甲醇选择性为2%。催化剂连续反应稳定,反应连续进行100h转化率下降在10%以内。
[0076]
合成实施例2
[0077]
1.在固定床反应器中加入上述制备实施例2中制备的催化剂2,氢气气氛下升温至300℃并保持3h后降至180℃。
[0078]
2.将苯与戊酸(摩尔比1:2)分别用柱塞泵(空速1.0h-1
)通入预热器预热后进入固定床反应器中进行反应。
[0079]
3.反应产物经过冷凝和气液分离。
[0080]
经gc检测,反应20h时,苯的转化率为77%,产物中戊基苯选择性88%,1-苯基-1-戊酮选择性3%,1-苯基-1-戊醇选择性4%。
[0081]
4.催化剂连续反应稳定,反应连续进行90h转化率下降在7%以内。
[0082]
合成实施例3
[0083]
1.在固定床反应器中加入上述制备实施例3中制备的催化剂3,在氢气气氛下升温至350℃并保持3h后降至180℃。
[0084]
2.将苯与丁二酸酐(10wt%乙醇溶液)(摩尔比1:2)分别用柱塞泵(空速1.0h-1
)通入预热器预热后进入固定床反应器中进行反应。
[0085]
3.反应产物经过冷凝和气液分离。
[0086]
经gc检测,反应20h时,苯的转化率为74%,苯丁酸选择性87%,4-羰基苯丁酸选择性5%,4-羟基苯丁酸选择性3%。催化剂连续反应稳定,反应连续进行90h转化率下降在10%以内。
[0087]
对比实施例1
[0088]
1.在固定床反应器中加入上述对比制备实施例1中制备的对比催化剂1,氢气气氛下升温至300℃并保持3h后降至180℃。
[0089]
2.将苯甲醚与醋酸酐摩尔比1:2)分别用柱塞泵(空速1.0h-1
)通入预热器预热后进入固定床反应器中进行反应。
[0090]
3.反应产物经过冷凝和气液分离。
[0091]
经gc检测,反应6h时,苯甲醚的转化率为79%,产物中4-乙基苯甲醚选择性89%,2-乙基苯甲醚选择性2%,对甲氧基苯乙酮选择性1%,对甲氧基苯-α-甲基苯甲醇选择性为3%。该催化剂连续反应前8h转化率较稳定,8-20h转化率下降20%,20h之后转化率急剧下降,30h时失活,转化率降至30%以下。
[0092]
对比实施例2
[0093]
1.在固定床反应器中加入上述对比制备实施例2中制备的对比催化剂2,在氢气气氛下升温至350℃并保持3h后降至180℃。
[0094]
2.将苯甲醚与醋酸酐(摩尔比1:2)分别用柱塞泵(空速1.0h-1
)通入预热器预热后进入固定床反应器中进行反应。
[0095]
3.反应产物经过冷凝和气液分离。
[0096]
经gc检测,反应10h时,苯甲醚的转化率为88%,产物中对甲氧基苯乙酮选择性91%。该催化剂连续反应前15h活性较稳定,15-40h活性下降23%,40h后活性急剧下降,50h时失活,转化率降至30%以下。
[0097]
图2和图3分别示出上述合成实施例1和对比实施例1中的反应的稳定性(即,苯甲醚的转化率及4-乙基苯甲醚选择性随着时间的变化)。由上述合成实施例1-3及对比实施例1-2可以看出,未负载金属活性组分(对比实施例2)以及未使用等离子体处理(对比实施例1,图3)时催化剂的稳定性差,在40h以内均出现转化率下降的现象。在未负载金属的催化剂上得到的主要为芳香酮产物,在负载金属后得到的主要产物为烷基取代的芳烃。在负载金属及使用等离子体处理后(合成实施例1-3),催化剂的稳定性得到明显改善,在连续运行90h时未出现明显的失活现象(图2)。另外,反应结束后的催化剂的热重分析结果(图1)显示,催化剂1在反应过程中生成的积碳量要明显少于对比催化剂1。
[0098]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。