包含碳-嵌入的镍纳米粒子的材料、其生产方法和作为非均相催化剂的用途
背景技术:
1.本发明涉及一种材料,其包含其中分散有镍纳米粒子(nanoparticles)的非石墨化碳的颗粒(grains)。根据本发明的材料在多种化学反应中具有催化活性,并且可以通过简单的方法获得。
2.本发明的碳相主要是无定形的,并且并不显示为活性炭、炭黑、石墨、石墨化炭黑或次晶碳(paracrystalline carbon)。
3.现有技术
4.大量现有技术都是针对合成过渡金属纳米粒子,特别是包括具有催化活性的过渡金属纳米粒子。然而,由于纳米粒子本身不能用于大多数非均相催化过程,因此进行了进一步的努力以开发含有附着于合适的载体、基材或晶片上的过渡金属纳米粒子的材料。用于此目的的现有技术方法主要基于将金属前体浸渍或化学气相沉积在多孔或介孔载体上的(sietsma,jelle r.a.等:“highly active cobalt-on-silica catalysts for the fischer-tropsch synthesis obtained via a novel calcination procedure.”studies in surface science and catalysis(2007);van deelen,t.w.等:"assembly and activation of supported cobalt nanocrystal catalysts for the fischer
–
tropsch synthesis."chemical communications(2018).),或对金属物质使用定义明确的配体并施加高温处理(westerhaus,felix a.等:“heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes”nature chemistry(2013);banerjee,debasis等:“convenient and mild epoxidation of alkenes using heterogeneous oxide catalysts”angewandte chemie,international edition(2014).)。然而,发现纳米粒子和载体的相互作用带来了显著的限制(oschatz,m.等:“effects of calcination and activation conditions on ordered mesoporous carbon supported iron catalysts for production of lower olefins from synthesis gas”catalysis science&technology(2016).)。特别地,现有技术的方法不能产生表现出过渡金属纳米粒子/金属氧化物纳米粒子的高分散和均匀配位以及高金属含量的材料。事实上,大多数现有技术的过渡金属纳米粒子材料由于在较高金属浓度下金属粒子的聚集和相应的分散损失而表现出低于20重量%的相当低活性的金属浓度(hern
á
ndez mej
í
a,carlos,tom w.van deelen und krijn p de jong.“activity enhancement of cobalt catalysts by tuning metal-support interactions”nature communications(2018);oschatz,m.等:“effects of calcination and activation conditions on ordered mesoporous carbon supported iron catalysts for production of lower olefins from synthesis gas.”catalysis science&technology(2016))。鉴于表现出过渡金属纳米粒子/金属氧化物纳米粒子的高分散和均匀配位以及高金属含量的材料目前尚不可得,而此类特性被认为是理想的,为了获得具有高催化活性的材料,本领域需要提供此类材料及其制备方法。
5.本发明提供了表现出所需性能的材料和用于生产其的简便方法。
技术实现要素:
6.本发明涉及催化活性材料,其包含其中分散有镍纳米粒子的非石墨化碳的颗粒,
7.其中
8.所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,
9.所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,并且
10.所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内,
11.其中d
p
和d由本文所述的tgz-tem测量,
12.并且其中
13.d
p
、d和ω符合以下关系:
14.4.5d
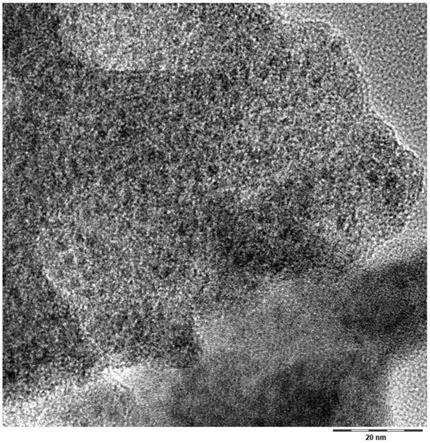
p
/ω》d≥0.25d
p
/ω。
15.根据本发明的材料可以通过包括以下步骤的方法获得:
16.(a)提供包含金属前体和有机碳源的水溶液,
17.其中所述金属前体包含一种有机的、至少部分水溶性的镍盐或多于一种的有机的、至少部分水溶性的镍盐的组合,并且
18.其中所述有机碳源是一种饱和的脂族二羧酸、三羧酸或多羧酸或多于一种的饱和的脂族二羧酸、三羧酸或多羧酸的组合,
19.(b)对所述金属前体和有机碳源的水溶液进行喷雾干燥或冷冻干燥,由此得到中间产物p,
20.(c)在200℃-380℃的温度范围内对所述中间产物p进行热处理。
21.作为本发明的基础的研究结果,发现通过组合以下步骤,可以由金属前体和有机碳源的水溶液获得其中分散有镍纳米粒子的非石墨化碳的颗粒:
22.(i)对所述水溶液进行喷雾干燥或冷冻干燥,
23.(ii)在中等温度下对由步骤(i)获得的中间体进行热处理。
24.发现最终产物在多种化学反应中表现出催化活性。在本发明的上下文中,任何降低化学反应的活化能并因此在特定温度下提高其速率而不被催化反应本身所消耗的材料或物质都被认为是催化活性的。
25.工艺条件的变化和所获得材料的检验揭示了本发明的工艺条件和材料性能。
26.发现,在玻璃烧杯中形成金属前体和有机碳源的水溶液,在干燥箱中缓慢干燥这些溶液过夜不会产生中间产物,所述中间产物可以通过在中等温度(moderate temperature)下热处理转变为其中分散有镍纳米粒子的非石墨化碳的颗粒。具体地,发现如果干燥过程进行得太慢,则多羧酸的大量分解和二氧化碳的形成过早开始,导致碳源的氧官能度(oxygen functionalities)早期损失。然而,氧官能度的早期损失似乎与金属组分的团聚和金属前体与碳源的分离相关,最终在碳基质内产生大尺寸金属簇的不规则分布。因此,不希望受理论约束,在干燥过程的部分过程中,含氧官能团的足够可用性对于以高度分散和规则的方式将金属前体固定在碳源中似乎是必不可少的。
27.此外发现,在低于200℃和高于380℃的温度下对中间产物p进行热处理不会产生其中分散有镍纳米粒子的根据本发明的非石墨化碳的颗粒。特别地发现,当选择用于热处理的温度太高时,根据本发明的所述非石墨化碳相本身的比例降低。然而,这些相被认为与有利的氢传导性(hydrogen conductivity)有关,而氢传导性对于涉及氢转化的有效催化反应是必要的。另一方面,如果选择用于热处理的温度太低或热处理的持续时间太短,则获得的碳相中的余氧水平太高,并且金属前体的还原仍然不完全,导致催化活性降低。
28.此外,应注意,鉴于现有技术,作为本发明方法的结果,本发明的非石墨化碳相的形成似乎是出人意料的。然而,不希望受理论约束,据信在随后的热处理之前,在中间产物p中以高度分散的方式存在高浓度的金属前体,促进了在本发明方法的低温条件下非石墨化碳的形成。
29.本发明的方法产生颗粒形式的非石墨化碳材料。非石墨化碳可以由技术人员使用tem分析来识别(参见p.w.albers,neutron scattering study of the terminating protons in the basic structural units of non-graphitizing and graphitizing carbons,carbon 109(2016),239-245,第241页,图1c)。
30.结合本发明获得的实验结果表明,通过本发明方法获得的材料的催化活性与其中表现出本发明特征的非石墨化碳的颗粒的含量密切相关。
31.通常,通过本发明的方法获得的非石墨化碳颗粒的90%呈现中等尺寸(moderate size),即直径在2μm至200μm之间。目前发现,通常,通过本发明的方法获得的那些中等尺寸的非石墨化碳的颗粒的超过95%含有分散在其中的镍纳米粒子,其符合4.5d
p
/ω》d≥0.25d
p
/ω(其中d
p
表示所述非石墨化碳颗粒中的镍纳米粒子的平均直径,d表示所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离,ω表示在所述非石墨化碳颗粒中金属的组合总质量分数)。本发明的方法通常产生这样的颗粒,其中只有非常小的颗粒部分和非常大的颗粒部分,即在2μm和200μm之间的中等尺寸范围之外的颗粒部分,含有大量的颗粒,其中镍纳米粒子不符合关系4.5d
p
/ω》d≥0.25d
p
/ω。因此,本发明的方法通常产生具有高含量的含镍纳米粒子的颗粒的材料,其中镍纳米粒子符合关系4.5d
p
/ω》d≥0.25d
p
/ω。然而,具有较低含量的这些颗粒的材料可以通过其他方法或用其他材料稀释获得,因此也包括在本发明中。
32.因此,在优选实施方案中,本发明涉及包含其中分散有镍纳米粒子的非石墨化碳的颗粒的催化活性材料,其中大于90%的中等尺寸的非石墨化碳颗粒中的镍纳米粒子,即直径在2μm和200μm之间的非石墨化碳颗粒符合关系:4.5d
p
/ω》d≥0.25d
p
/ω,并且其中,所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内。
33.在另一个优选实施方案中,本发明涉及包含其中分散有镍纳米粒子的非石墨化碳的颗粒的催化活性材料,其中大于95%的中等尺寸的非石墨化碳颗粒中的镍纳米粒子,即直径在2μm和200μm之间的非石墨化碳颗粒符合关系:4.5d
p
/ω》d≥0.25d
p
/ω,并且其中,所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内。
34.本发明的非石墨化碳材料中的镍纳米粒子主要由元素镍组成,但也可以含有例如氧化镍和/或掺杂金属和/或第二金属(secondary metal)。
35.计算机辅助分析tem图片(tem=透射电子显微镜)与来自degussa的tgz方法相结合,可以确定单个镍纳米粒子的直径以及其集合(sets)的统计测量(参见parker et al.“the effect of particle size,morphology and support on the formation of palladium hydride in commercial catalysts”chemical science,2019,10,480)。
36.在本发明的上下文中,镍纳米粒子的平均直径d
p
和平均距离d通过tgz-tem方法测量,如下所述:
37.1.样品制备
38.在大多数情况下,待测样品是粉末。
39.通常在超声波作用下,将粉末分散在溶剂中。超声波的应用将团聚体分解成聚集体,其结果是聚集体的分布,而不是聚集体和团聚体的混合物。然后用微型移液管将液滴滴到滤纸上的涂膜网孔上。过量的液体很快通过滤纸被吸出,从而通过干燥过程防止团聚体形成。悬浮颗粒不能太密集,因为通过颗粒的接触和重叠不能清楚地看到纳米粒子的形状和轮廓。最佳稀释度必须通过稀释系列的试验来确定。
40.通常,可以说制备物的类型对初级纳米粒子尺寸评价的结果几乎没有任何影响。
41.2.测试的表现
42.基于tem图像表征的单个纳米粒子必须以足够清晰的轮廓成像。
43.在tem图像上的纳米粒子分布不太密集,几乎没有重叠,或者粒子之间尽可能分离,这有利于tgz3上的测量,但不影响测量结果。
44.在检查tem制备的各个图像部分之后,相应地选择合适的区域。应该注意,对于各个样品,小、中、大纳米粒子的比例是代表性的和表征性的,操作者没有给出小或大粒子的选择性偏好。
45.待测量的初级纳米粒子的总数取决于初级纳米粒子尺寸的分散范围:分散范围越大,必须测量的粒子越多,以获得足够的统计报表(statistical statement)。对于金属催化剂,测定了约1500个单个粒子。对于所有的tgz分析,使用在100kev下操作的配备有ccd照相机的校准的hitachi h-7500场透射电子显微镜。
46.3.测量程序说明
47.测量程序是根据carl zeiss的tgz3手册(“(particle size analyser)tgz3”;fa.carl zeiss手册)进行的。
48.4.测量数据处理
49.测量数据处理的详细描述在(f.endter u.h.gebauer,“optik(optics)”13(1956),97)和(k.seibold und m.voll,“distribution function for describing the particle size distribution of soot and pyrogenic oxides”.chemiker-zeitung,102(1978),nr.4,131-135)中给出。
50.统计总结以报告的形式编辑。详细的统计描述在(lothar sachs,“statistical methods”,5.auflage,springer-verlag,berlin(1982))中给出。
51.5.结果的评价和展示
52.a)粒子总数(n)
53.b)对每个样品1500个分离的纳米粒子评价粒度分布q0(x)和q3(x)
54.c)粒径dn,平均粒径(dn)
[0055][0056]
ni=直径为di的粒子数
[0057]
d)矩形平面上的平均距离d
[0058][0059]
a、b=矩形平面的长、宽
[0060]
x、y、x*、y*=粒子坐标。
[0061]
金属的组合总质量分数ω定义为镍、所有掺杂金属和第二金属的组合总质量占所考虑材料总质量的分数:ω=(m(镍) m(掺杂金属) m(第二金属))/m(材料);其中m(镍)=以元素镍本身的形式和/或以镍的任何化合物的形式包含在材料中的元素形式的镍的总质量,m(掺杂金属)=以元素掺杂金属本身的形式和/或以掺杂金属的任何化合物的形式包含在材料中的元素形式的所有掺杂金属的组合总质量,m(第二金属)=以元素第二金属本身的形式和/或以任何第二金属化合物的形式包含在材料中的所有元素形式的第二金属的组合总质量,m(材料)=所考虑的材料的总质量。
[0062]
金属的组合总质量分数ω可以用各种元素定量分析方法,特别是xrf(x射线荧光)和icp-aes(电感耦合等离子体原子发射光谱)来测定。
[0063]
适当地选择根据本发明的方法中的条件可以控制所获得的材料中金属的组合总质量分数ω:
[0064]
与在步骤(a)中提供具有较低金属含量(镍、掺杂金属和第二金属的组合)的溶液的方法相比,在步骤(a)中提供具有高金属含量的溶液的方法产生具有更高的金属的组合总质量分数ω的材料。
[0065]
在200℃至380℃范围内的高温下进行步骤(c)的热处理的方法比在较低温度下进行步骤(c)的热处理的方法获得具有更高的金属的组合总质量分数ω。
[0066]
本发明的方法产生粒状材料。该材料的单个粒子的尺寸及其集合的统计测量可以通过本领域技术人员公知的激光衍射分析(例如cilas 1190系列)来确定。
[0067]
通常,本发明的方法产生具有以下粒度分布的粒状材料:d10=5μm,d50=40μm,d90=150μm。
[0068]
鉴于发现通过根据本发明的方法获得的材料非常适合生产成形催化剂的事实,在优选实施方案中,本发明涉及催化活性材料,其包含其中分散有镍纳米粒子的非石墨化碳的颗粒,
[0069]
其中
[0070]
所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,
[0071]
所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,并且
[0072]
所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内,
[0073]
并且其中
[0074]dp
、d和ω符合以下关系:
[0075]
4.5d
p
/ω》d≥0.25d
p
/ω,
[0076]
并且其中
[0077]
所述非石墨化碳颗粒显示出以下粒度分布:d10=5μm,d50=40μm,d90=150μm。
[0078]
根据本发明的材料可以应用于氮的存在有害的情况。因此,在优选实施方案中,本发明涉及根据本发明的材料,其中氮的总质量分数小于材料总质量的1重量%。
[0079]
实验结果表明,具有相对较小镍纳米粒子的材料可能表现出特别有吸引力的催化性能。因此,在优选的实施方案中,本发明涉及根据本发明的材料,其中d
p
在2nm-16nm的范围内。在特别优选的实施方案中,本发明涉及根据本发明的材料,其中d
p
在4nm-12nm的范围内。
[0080]
如实验结果所示,掺杂金属的加入影响了本发明的材料的催化活性。因此,在优选实施方案中,本发明涉及根据本发明的材料,其中所述镍纳米粒子已经掺杂有掺杂金属,并且其中所述掺杂金属选自na(钠)、k(钾)、v(钒)、la(镧)、mg(镁)、ce(铈)或其混合物,并且其中所述材料的摩尔比rdm=n(镍):n(掺杂金属)在2-1000范围内。
[0081]
在特别优选的实施方案中,本发明涉及根据本发明的材料,其中所述镍纳米粒子已经掺杂有掺杂金属,并且其中所述掺杂金属选自na(钠)、k(钾)、v(钒)、la(镧)、mg(镁)、ce(铈)或它们的混合物,并且其中所述材料的摩尔比rdm=n(镍):n(掺杂金属)在4-500的范围内。
[0082]
在另一个优选实施方案中,本发明涉及根据本发明的材料,其中所述镍纳米粒子已经与第二金属组合,并且其中所述第二金属选自第1组或第2组,
[0083]
第1组定义为:mo(钼)或w(钨)或其混合物,且
[0084]
第2组定义为:co(钴)或cu(铜)或mn(锰)或其混合物,
[0085]
并且其中所述材料的摩尔比rsm=n(镍):n(第二金属)在1-50的范围内。
[0086]
本发明还涉及生产本发明的材料的方法。如上所述,发现下述两个方法步骤的组合是至关重要的:
[0087]
(i)对金属前体和有机碳源的水溶液进行喷雾干燥或冷冻干燥,和
[0088]
(ii)在中等温度下对所得中间体进行热处理。
[0089]
因此,在另一方面,本发明进一步涉及用于生产根据本发明的材料的方法,其包括以下步骤:
[0090]
(a)提供包含金属前体和有机碳源的水溶液,
[0091]
其中所述金属前体包含一种有机的、至少部分水溶性的镍盐或多于一种的有机的、至少部分水溶性的镍盐的组合,且
[0092]
其中所述有机碳源是一种饱和的脂族二羧酸、三羧酸或多羧酸或多于一种的饱和的脂族二羧酸、三羧酸或多羧酸的组合,
[0093]
(b)对所述金属前体和有机碳源的水溶液进行喷雾干燥或冷冻干燥,由此得到中间产物p,
[0094]
(c)在200℃至380℃的温度范围内对中间产物p进行热处理。
[0095]
每个方法步骤可以以间歇或连续的形式进行。
[0096]
在另一方面,本发明还涉及可通过本发明的方法获得的材料。
[0097]
如上所述,本发明的材料的形成需要喷雾干燥或冷冻干燥与在中等温度下的适当热处理的组合。因此,似乎可以合理地认为只有存在于溶液中的材料,即在该方法的步骤(a)中提供的溶液中以溶解形式存在的材料,才能转化为根据本发明的材料。然而,固体形式的未溶解物质可以悬浮在步骤(a)中提供的溶液中,只要它不干扰形成本发明材料的方法即可。这些固体,其可以例如源自未溶解的金属前体或有机碳源,可以在本发明的方法的步骤(c)后获得的固体产物中形成本发明的材料的固体稀释物。类似地,有机溶剂可以在步骤(a)中提供的溶液中溶解或乳化,只要其存在不干扰形成本发明的材料的方法即可。然而,为了避免干扰形成本发明的材料的方法,在优选实施方案中,本发明的方法是用步骤(a)中提供的水溶液进行的,该水溶液不含固体形式的未溶解物质,也不含有机溶剂。
[0098]
如果不使用掺杂金属和第二金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种有机的、至少部分水溶性的镍盐或多于一种的有机的、至少部分水溶性的镍盐的组合。在本发明中,如果盐的至少一部分在该方法中使用的条件下溶解在步骤(a)中提供的水溶液中,则认为盐至少部分是水溶性的。优选地,如果不使用掺杂金属且不使用第二金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多于一种有机镍盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0099]
在本发明的优选实施方案中,不使用掺杂金属和第二金属。
[0100]
在本发明的另一个优选实施方案中,使用掺杂金属,但不使用第二金属。
[0101]
如果使用掺杂金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机的、至少部分水溶性的镍盐与一种或多种掺杂金属的一种或多种有机的、至少部分水溶性的盐的组合。优选地,如果使用掺杂金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机镍盐与一种或多种掺杂金属的一种或多种有机盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0102]
如果使用第二金属,则在本发明方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机的、至少部分水溶性的镍盐与一种或多种第二金属的一种或多种有机的、至少部分水溶性的盐的组合。优选地,如果使用第二金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机镍盐与一种或多种第二金属的一种或多种有机盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0103]
如果使用掺杂金属和第二金属,在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机的、至少部分水溶性的镍盐与一种或多种掺杂金属的一种或多种有机的、至少部分水溶性的盐和一种或多种第二金属的一种或多种有机的、至少部分水溶性的盐的组合。优选地,如果使用掺杂金属和第二金属,在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机镍盐与一种或多种掺杂金属的一种或多种有机盐和一种或多种第二金属的一种或多种有机的、至少部分水溶性的盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0104]
在本发明的方法的步骤(a)中提供的溶液中,金属前体的优选有机阴离子是乙酸根、碳酸根、草酸根、柠檬酸根、丙二酸根、酒石酸根和戊二酸根。如果不需要避免氮,则硝酸根是步骤(a)中提供的溶液中金属前体的另一个优选阴离子。
[0105]
饱和的脂族二羧酸、三羧酸或多羧酸,单独或作为混合物的一部分,可以用作步骤
(a)中提供的水溶液的有机碳源,只要其支持本发明的材料的形成。在优选实施方案中,丙二酸、戊二酸、柠檬酸或其混合物用作本发明的方法的步骤(a)中提供的水溶液的有机碳源。在本发明的特别优选的实施方案中,柠檬酸用作本发明的方法的步骤(a)中提供的水溶液的有机碳源。
[0106]
在本发明方法的步骤(b)中,对在步骤(a)中提供的水溶液进行喷雾干燥或冷冻干燥。由此获得的产物在本发明的上下文中称为中间产物p。喷雾干燥和冷冻干燥的工艺参数可以在宽范围内变化,只要干燥过程不间断进行,并且中间产物p所表现出的水和有机溶剂的组合含量低于10重量%即可。在本发明的优选实施方案中,在本发明的方法的步骤(b)中对步骤(a)中提供的水溶液进行喷雾干燥。
[0107]
根据本发明的方法的步骤(c)的热处理是在规定的温度条件和惰性气体气氛(如氮气或空气)下进行的。用于此目的的多种合适的炉可商购获得。在优选实施方案中,热处理在惰性气体气氛(例如氮气)下进行。热处理过程中的加热速率应足够小,以使热量均匀分布,即通常小于15k/min,优选小于10k/min,特别优选小于5k/min。中间产物p在200℃-380℃的温度范围内进行热处理。在本发明的优选实施方案中,中间产物p在255℃-375℃的温度范围内进行热处理。在特别优选的实施方案中,中间产物p在300℃-350℃的温度范围内进行热处理。通常,对中间产物p进行1-4小时的热处理,但延长或缩短时间间隔的热处理也可以起作用。当确定热处理的持续时间时,不考虑加热和冷却间隔。在优选实施方案中,对所述中间产物p进行1-4小时的热处理。
[0108]
如上所述,根据本发明的材料表现出催化活性。因此,在另一方面,本发明进一步涉及本发明的材料作为催化剂的用途。
[0109]
根据本发明的材料例如可用作有机化合物的液相氢化的催化剂,所述有机化合物特别是不饱和化合物如烯烃、不饱和羧酸和不饱和醇、炔烃、醛和酮、酯和亚胺、硝基化合物和腈以及芳基化合物。此外,根据本发明的材料是用于羰基化合物的还原胺化的活性非常强的催化剂。因此,在另一方面,本发明进一步涉及本发明的材料作为用于有机化合物的氢化、羰基化合物的还原胺化和/或有机化合物的加氢甲酰化的催化剂的用途。
[0110]
根据本发明的材料还可以用作用氢气将一氧化碳、二氧化碳或其混合物转化为饱和及不饱和的烃或其混合物中的催化剂。因此,在另一方面,本发明还涉及本发明的材料作为用氢气将一氧化碳、二氧化碳或其混合物转化为甲烷的催化剂的用途。
[0111]
根据本发明的材料可以作为未改性形式的催化剂使用,或者可以通过本领域技术人员所公知的成型方法(例如压片、造粒、挤出、涂覆、3d打印)转化成催化剂体。
实施例
[0112]
实施例ni a、b-碳嵌入的ni纳米粒子(carbon embedded ni-nanoparticles)的制备
[0113]
通过在室温下连续搅拌,将14.4g柠檬酸(特纯(puriss),sigma aldrich)溶解于75ml去离子水中,制备碳嵌入的ni纳米粒子。在第二个烧杯中,在室温下连续搅拌下,将18.7g乙酸镍(ii)四水合物(ni(ch3coo)2*4h2o,sigma aldrich)溶解于75ml去离子水中。将乙酸镍溶液缓慢加入到柠檬酸溶液中,并在室温下再搅拌30分钟。用常规小型喷雾干燥机(b
ü
chi,mini spray dryer b-290)喷雾干燥所得溶液,所述常规小型喷雾干燥机具有220
℃的恒定入口温度、120℃的出口温度和20%的泵速。将获得的粉末分成质量相同的两部分进行最终的热处理。
[0114]
将第一个样品在氮气气氛下的管式炉中以180分钟升温(ramp)至300℃进行热处理,其中将温度再保持4小时,然后自然冷却。得到的催化剂粉末标记为nicat.1a。
[0115]
将第二个样品在氮气气氛下以类似的方式热处理。在180分钟内将样品加热至350℃,其中将温度保持4小时,然后自然冷却。得到的催化剂粉末标记为nicat.1b。
[0116]
使用在100kev下操作的配备有ccd照相机的校准的hitachi h-7500场透射电子显微镜,通过xrf(x射线荧光)和tgz分析,确定了这些材料具有以下特征:
[0117]
idd
p
ωdnicat.1a9.5nm0.5315nmnicat.1b11nm0.5812nm
[0118]
比较例
[0119]
为了比较,在传统的vulcan xc72r碳载体上,用初期湿浸渍法(incipient wetness impregnation)制备了镍含量为20%的高负载催化剂,并将其标记为nicat.ref。
[0120]
使用在100kev下操作的配备有ccd照相机的校准的hitachi h-7500场透射电子显微镜,通过xrf(x射线荧光)和tgz分析,确定了这些材料具有以下特征:
[0121]
idd
p
ωdnicat.ref67nm0.20n.d.*
[0122]
催化活性试验
[0123]
用200mg催化剂和5mmol底物在5ml甲醇中分批进行试验,以测定材料的催化活性和选择性。对于所有实验,将高压釜加热至所需的反应温度,并在50bar的恒定氢气压力下搅拌。过滤反应产物并通过gc-ms进行分析。
[0124]
i.n-亚苄基-苄胺的氢化
[0125][0126]
idcat.id持续时间h温度t℃反应物%产物%副产物%1nicat.1a20.00100.002.4088.98.702nicat.1b20.00100.001.7095.03.303nicat.ref20.00100.0064.023.612.4
[0127]
ii.巴豆酸甲酯氢化成丁酸甲酯
[0128][0129]
idcat.id持续时间h温度t℃反应物%产物%4nicat.1a20.0080.000.00100.005nicat.1b20.0080.000.00100.006nicat.ref20.0080.001.198.9
[0130]
iii.十二腈(dodecannitrile)的氢化
[0131][0132]
idcat.id持续时间h温度t℃反应物%产物%副产物%7nicat.1a20.0080.0065.528.85.68nicat.1b20.0080.00073.027.09nicat.ref20.0080.0072.520.17.4
[0133]
iv.乙酰基萘的氢化
[0134][0135]
idcat.id持续时间h温度t℃反应物%产物%副产物%10nicat.1a16.0080.000.00100.000.0011nicat.1b16.0080.000.00100.000.0012nicat.ref20.0080.000.00100.000.00
背景技术:
1.本发明涉及一种材料,其包含其中分散有镍纳米粒子(nanoparticles)的非石墨化碳的颗粒(grains)。根据本发明的材料在多种化学反应中具有催化活性,并且可以通过简单的方法获得。
2.本发明的碳相主要是无定形的,并且并不显示为活性炭、炭黑、石墨、石墨化炭黑或次晶碳(paracrystalline carbon)。
3.现有技术
4.大量现有技术都是针对合成过渡金属纳米粒子,特别是包括具有催化活性的过渡金属纳米粒子。然而,由于纳米粒子本身不能用于大多数非均相催化过程,因此进行了进一步的努力以开发含有附着于合适的载体、基材或晶片上的过渡金属纳米粒子的材料。用于此目的的现有技术方法主要基于将金属前体浸渍或化学气相沉积在多孔或介孔载体上的(sietsma,jelle r.a.等:“highly active cobalt-on-silica catalysts for the fischer-tropsch synthesis obtained via a novel calcination procedure.”studies in surface science and catalysis(2007);van deelen,t.w.等:"assembly and activation of supported cobalt nanocrystal catalysts for the fischer
–
tropsch synthesis."chemical communications(2018).),或对金属物质使用定义明确的配体并施加高温处理(westerhaus,felix a.等:“heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes”nature chemistry(2013);banerjee,debasis等:“convenient and mild epoxidation of alkenes using heterogeneous oxide catalysts”angewandte chemie,international edition(2014).)。然而,发现纳米粒子和载体的相互作用带来了显著的限制(oschatz,m.等:“effects of calcination and activation conditions on ordered mesoporous carbon supported iron catalysts for production of lower olefins from synthesis gas”catalysis science&technology(2016).)。特别地,现有技术的方法不能产生表现出过渡金属纳米粒子/金属氧化物纳米粒子的高分散和均匀配位以及高金属含量的材料。事实上,大多数现有技术的过渡金属纳米粒子材料由于在较高金属浓度下金属粒子的聚集和相应的分散损失而表现出低于20重量%的相当低活性的金属浓度(hern
á
ndez mej
í
a,carlos,tom w.van deelen und krijn p de jong.“activity enhancement of cobalt catalysts by tuning metal-support interactions”nature communications(2018);oschatz,m.等:“effects of calcination and activation conditions on ordered mesoporous carbon supported iron catalysts for production of lower olefins from synthesis gas.”catalysis science&technology(2016))。鉴于表现出过渡金属纳米粒子/金属氧化物纳米粒子的高分散和均匀配位以及高金属含量的材料目前尚不可得,而此类特性被认为是理想的,为了获得具有高催化活性的材料,本领域需要提供此类材料及其制备方法。
5.本发明提供了表现出所需性能的材料和用于生产其的简便方法。
技术实现要素:
6.本发明涉及催化活性材料,其包含其中分散有镍纳米粒子的非石墨化碳的颗粒,
7.其中
8.所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,
9.所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,并且
10.所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内,
11.其中d
p
和d由本文所述的tgz-tem测量,
12.并且其中
13.d
p
、d和ω符合以下关系:
14.4.5d
p
/ω》d≥0.25d
p
/ω。
15.根据本发明的材料可以通过包括以下步骤的方法获得:
16.(a)提供包含金属前体和有机碳源的水溶液,
17.其中所述金属前体包含一种有机的、至少部分水溶性的镍盐或多于一种的有机的、至少部分水溶性的镍盐的组合,并且
18.其中所述有机碳源是一种饱和的脂族二羧酸、三羧酸或多羧酸或多于一种的饱和的脂族二羧酸、三羧酸或多羧酸的组合,
19.(b)对所述金属前体和有机碳源的水溶液进行喷雾干燥或冷冻干燥,由此得到中间产物p,
20.(c)在200℃-380℃的温度范围内对所述中间产物p进行热处理。
21.作为本发明的基础的研究结果,发现通过组合以下步骤,可以由金属前体和有机碳源的水溶液获得其中分散有镍纳米粒子的非石墨化碳的颗粒:
22.(i)对所述水溶液进行喷雾干燥或冷冻干燥,
23.(ii)在中等温度下对由步骤(i)获得的中间体进行热处理。
24.发现最终产物在多种化学反应中表现出催化活性。在本发明的上下文中,任何降低化学反应的活化能并因此在特定温度下提高其速率而不被催化反应本身所消耗的材料或物质都被认为是催化活性的。
25.工艺条件的变化和所获得材料的检验揭示了本发明的工艺条件和材料性能。
26.发现,在玻璃烧杯中形成金属前体和有机碳源的水溶液,在干燥箱中缓慢干燥这些溶液过夜不会产生中间产物,所述中间产物可以通过在中等温度(moderate temperature)下热处理转变为其中分散有镍纳米粒子的非石墨化碳的颗粒。具体地,发现如果干燥过程进行得太慢,则多羧酸的大量分解和二氧化碳的形成过早开始,导致碳源的氧官能度(oxygen functionalities)早期损失。然而,氧官能度的早期损失似乎与金属组分的团聚和金属前体与碳源的分离相关,最终在碳基质内产生大尺寸金属簇的不规则分布。因此,不希望受理论约束,在干燥过程的部分过程中,含氧官能团的足够可用性对于以高度分散和规则的方式将金属前体固定在碳源中似乎是必不可少的。
27.此外发现,在低于200℃和高于380℃的温度下对中间产物p进行热处理不会产生其中分散有镍纳米粒子的根据本发明的非石墨化碳的颗粒。特别地发现,当选择用于热处理的温度太高时,根据本发明的所述非石墨化碳相本身的比例降低。然而,这些相被认为与有利的氢传导性(hydrogen conductivity)有关,而氢传导性对于涉及氢转化的有效催化反应是必要的。另一方面,如果选择用于热处理的温度太低或热处理的持续时间太短,则获得的碳相中的余氧水平太高,并且金属前体的还原仍然不完全,导致催化活性降低。
28.此外,应注意,鉴于现有技术,作为本发明方法的结果,本发明的非石墨化碳相的形成似乎是出人意料的。然而,不希望受理论约束,据信在随后的热处理之前,在中间产物p中以高度分散的方式存在高浓度的金属前体,促进了在本发明方法的低温条件下非石墨化碳的形成。
29.本发明的方法产生颗粒形式的非石墨化碳材料。非石墨化碳可以由技术人员使用tem分析来识别(参见p.w.albers,neutron scattering study of the terminating protons in the basic structural units of non-graphitizing and graphitizing carbons,carbon 109(2016),239-245,第241页,图1c)。
30.结合本发明获得的实验结果表明,通过本发明方法获得的材料的催化活性与其中表现出本发明特征的非石墨化碳的颗粒的含量密切相关。
31.通常,通过本发明的方法获得的非石墨化碳颗粒的90%呈现中等尺寸(moderate size),即直径在2μm至200μm之间。目前发现,通常,通过本发明的方法获得的那些中等尺寸的非石墨化碳的颗粒的超过95%含有分散在其中的镍纳米粒子,其符合4.5d
p
/ω》d≥0.25d
p
/ω(其中d
p
表示所述非石墨化碳颗粒中的镍纳米粒子的平均直径,d表示所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离,ω表示在所述非石墨化碳颗粒中金属的组合总质量分数)。本发明的方法通常产生这样的颗粒,其中只有非常小的颗粒部分和非常大的颗粒部分,即在2μm和200μm之间的中等尺寸范围之外的颗粒部分,含有大量的颗粒,其中镍纳米粒子不符合关系4.5d
p
/ω》d≥0.25d
p
/ω。因此,本发明的方法通常产生具有高含量的含镍纳米粒子的颗粒的材料,其中镍纳米粒子符合关系4.5d
p
/ω》d≥0.25d
p
/ω。然而,具有较低含量的这些颗粒的材料可以通过其他方法或用其他材料稀释获得,因此也包括在本发明中。
32.因此,在优选实施方案中,本发明涉及包含其中分散有镍纳米粒子的非石墨化碳的颗粒的催化活性材料,其中大于90%的中等尺寸的非石墨化碳颗粒中的镍纳米粒子,即直径在2μm和200μm之间的非石墨化碳颗粒符合关系:4.5d
p
/ω》d≥0.25d
p
/ω,并且其中,所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内。
33.在另一个优选实施方案中,本发明涉及包含其中分散有镍纳米粒子的非石墨化碳的颗粒的催化活性材料,其中大于95%的中等尺寸的非石墨化碳颗粒中的镍纳米粒子,即直径在2μm和200μm之间的非石墨化碳颗粒符合关系:4.5d
p
/ω》d≥0.25d
p
/ω,并且其中,所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内。
34.本发明的非石墨化碳材料中的镍纳米粒子主要由元素镍组成,但也可以含有例如氧化镍和/或掺杂金属和/或第二金属(secondary metal)。
35.计算机辅助分析tem图片(tem=透射电子显微镜)与来自degussa的tgz方法相结合,可以确定单个镍纳米粒子的直径以及其集合(sets)的统计测量(参见parker et al.“the effect of particle size,morphology and support on the formation of palladium hydride in commercial catalysts”chemical science,2019,10,480)。
36.在本发明的上下文中,镍纳米粒子的平均直径d
p
和平均距离d通过tgz-tem方法测量,如下所述:
37.1.样品制备
38.在大多数情况下,待测样品是粉末。
39.通常在超声波作用下,将粉末分散在溶剂中。超声波的应用将团聚体分解成聚集体,其结果是聚集体的分布,而不是聚集体和团聚体的混合物。然后用微型移液管将液滴滴到滤纸上的涂膜网孔上。过量的液体很快通过滤纸被吸出,从而通过干燥过程防止团聚体形成。悬浮颗粒不能太密集,因为通过颗粒的接触和重叠不能清楚地看到纳米粒子的形状和轮廓。最佳稀释度必须通过稀释系列的试验来确定。
40.通常,可以说制备物的类型对初级纳米粒子尺寸评价的结果几乎没有任何影响。
41.2.测试的表现
42.基于tem图像表征的单个纳米粒子必须以足够清晰的轮廓成像。
43.在tem图像上的纳米粒子分布不太密集,几乎没有重叠,或者粒子之间尽可能分离,这有利于tgz3上的测量,但不影响测量结果。
44.在检查tem制备的各个图像部分之后,相应地选择合适的区域。应该注意,对于各个样品,小、中、大纳米粒子的比例是代表性的和表征性的,操作者没有给出小或大粒子的选择性偏好。
45.待测量的初级纳米粒子的总数取决于初级纳米粒子尺寸的分散范围:分散范围越大,必须测量的粒子越多,以获得足够的统计报表(statistical statement)。对于金属催化剂,测定了约1500个单个粒子。对于所有的tgz分析,使用在100kev下操作的配备有ccd照相机的校准的hitachi h-7500场透射电子显微镜。
46.3.测量程序说明
47.测量程序是根据carl zeiss的tgz3手册(“(particle size analyser)tgz3”;fa.carl zeiss手册)进行的。
48.4.测量数据处理
49.测量数据处理的详细描述在(f.endter u.h.gebauer,“optik(optics)”13(1956),97)和(k.seibold und m.voll,“distribution function for describing the particle size distribution of soot and pyrogenic oxides”.chemiker-zeitung,102(1978),nr.4,131-135)中给出。
50.统计总结以报告的形式编辑。详细的统计描述在(lothar sachs,“statistical methods”,5.auflage,springer-verlag,berlin(1982))中给出。
51.5.结果的评价和展示
52.a)粒子总数(n)
53.b)对每个样品1500个分离的纳米粒子评价粒度分布q0(x)和q3(x)
54.c)粒径dn,平均粒径(dn)
[0055][0056]
ni=直径为di的粒子数
[0057]
d)矩形平面上的平均距离d
[0058][0059]
a、b=矩形平面的长、宽
[0060]
x、y、x*、y*=粒子坐标。
[0061]
金属的组合总质量分数ω定义为镍、所有掺杂金属和第二金属的组合总质量占所考虑材料总质量的分数:ω=(m(镍) m(掺杂金属) m(第二金属))/m(材料);其中m(镍)=以元素镍本身的形式和/或以镍的任何化合物的形式包含在材料中的元素形式的镍的总质量,m(掺杂金属)=以元素掺杂金属本身的形式和/或以掺杂金属的任何化合物的形式包含在材料中的元素形式的所有掺杂金属的组合总质量,m(第二金属)=以元素第二金属本身的形式和/或以任何第二金属化合物的形式包含在材料中的所有元素形式的第二金属的组合总质量,m(材料)=所考虑的材料的总质量。
[0062]
金属的组合总质量分数ω可以用各种元素定量分析方法,特别是xrf(x射线荧光)和icp-aes(电感耦合等离子体原子发射光谱)来测定。
[0063]
适当地选择根据本发明的方法中的条件可以控制所获得的材料中金属的组合总质量分数ω:
[0064]
与在步骤(a)中提供具有较低金属含量(镍、掺杂金属和第二金属的组合)的溶液的方法相比,在步骤(a)中提供具有高金属含量的溶液的方法产生具有更高的金属的组合总质量分数ω的材料。
[0065]
在200℃至380℃范围内的高温下进行步骤(c)的热处理的方法比在较低温度下进行步骤(c)的热处理的方法获得具有更高的金属的组合总质量分数ω。
[0066]
本发明的方法产生粒状材料。该材料的单个粒子的尺寸及其集合的统计测量可以通过本领域技术人员公知的激光衍射分析(例如cilas 1190系列)来确定。
[0067]
通常,本发明的方法产生具有以下粒度分布的粒状材料:d10=5μm,d50=40μm,d90=150μm。
[0068]
鉴于发现通过根据本发明的方法获得的材料非常适合生产成形催化剂的事实,在优选实施方案中,本发明涉及催化活性材料,其包含其中分散有镍纳米粒子的非石墨化碳的颗粒,
[0069]
其中
[0070]
所述非石墨化碳颗粒中的镍纳米粒子的平均直径d
p
在1nm-20nm的范围内,
[0071]
所述非石墨化碳颗粒中的镍纳米粒子之间的平均距离d在2nm-150nm的范围内,并且
[0072]
所述非石墨化碳颗粒中金属的组合总质量分数ω在所述非石墨化碳颗粒的总质量的30重量%-70重量%的范围内,
[0073]
并且其中
[0074]dp
、d和ω符合以下关系:
[0075]
4.5d
p
/ω》d≥0.25d
p
/ω,
[0076]
并且其中
[0077]
所述非石墨化碳颗粒显示出以下粒度分布:d10=5μm,d50=40μm,d90=150μm。
[0078]
根据本发明的材料可以应用于氮的存在有害的情况。因此,在优选实施方案中,本发明涉及根据本发明的材料,其中氮的总质量分数小于材料总质量的1重量%。
[0079]
实验结果表明,具有相对较小镍纳米粒子的材料可能表现出特别有吸引力的催化性能。因此,在优选的实施方案中,本发明涉及根据本发明的材料,其中d
p
在2nm-16nm的范围内。在特别优选的实施方案中,本发明涉及根据本发明的材料,其中d
p
在4nm-12nm的范围内。
[0080]
如实验结果所示,掺杂金属的加入影响了本发明的材料的催化活性。因此,在优选实施方案中,本发明涉及根据本发明的材料,其中所述镍纳米粒子已经掺杂有掺杂金属,并且其中所述掺杂金属选自na(钠)、k(钾)、v(钒)、la(镧)、mg(镁)、ce(铈)或其混合物,并且其中所述材料的摩尔比rdm=n(镍):n(掺杂金属)在2-1000范围内。
[0081]
在特别优选的实施方案中,本发明涉及根据本发明的材料,其中所述镍纳米粒子已经掺杂有掺杂金属,并且其中所述掺杂金属选自na(钠)、k(钾)、v(钒)、la(镧)、mg(镁)、ce(铈)或它们的混合物,并且其中所述材料的摩尔比rdm=n(镍):n(掺杂金属)在4-500的范围内。
[0082]
在另一个优选实施方案中,本发明涉及根据本发明的材料,其中所述镍纳米粒子已经与第二金属组合,并且其中所述第二金属选自第1组或第2组,
[0083]
第1组定义为:mo(钼)或w(钨)或其混合物,且
[0084]
第2组定义为:co(钴)或cu(铜)或mn(锰)或其混合物,
[0085]
并且其中所述材料的摩尔比rsm=n(镍):n(第二金属)在1-50的范围内。
[0086]
本发明还涉及生产本发明的材料的方法。如上所述,发现下述两个方法步骤的组合是至关重要的:
[0087]
(i)对金属前体和有机碳源的水溶液进行喷雾干燥或冷冻干燥,和
[0088]
(ii)在中等温度下对所得中间体进行热处理。
[0089]
因此,在另一方面,本发明进一步涉及用于生产根据本发明的材料的方法,其包括以下步骤:
[0090]
(a)提供包含金属前体和有机碳源的水溶液,
[0091]
其中所述金属前体包含一种有机的、至少部分水溶性的镍盐或多于一种的有机的、至少部分水溶性的镍盐的组合,且
[0092]
其中所述有机碳源是一种饱和的脂族二羧酸、三羧酸或多羧酸或多于一种的饱和的脂族二羧酸、三羧酸或多羧酸的组合,
[0093]
(b)对所述金属前体和有机碳源的水溶液进行喷雾干燥或冷冻干燥,由此得到中间产物p,
[0094]
(c)在200℃至380℃的温度范围内对中间产物p进行热处理。
[0095]
每个方法步骤可以以间歇或连续的形式进行。
[0096]
在另一方面,本发明还涉及可通过本发明的方法获得的材料。
[0097]
如上所述,本发明的材料的形成需要喷雾干燥或冷冻干燥与在中等温度下的适当热处理的组合。因此,似乎可以合理地认为只有存在于溶液中的材料,即在该方法的步骤(a)中提供的溶液中以溶解形式存在的材料,才能转化为根据本发明的材料。然而,固体形式的未溶解物质可以悬浮在步骤(a)中提供的溶液中,只要它不干扰形成本发明材料的方法即可。这些固体,其可以例如源自未溶解的金属前体或有机碳源,可以在本发明的方法的步骤(c)后获得的固体产物中形成本发明的材料的固体稀释物。类似地,有机溶剂可以在步骤(a)中提供的溶液中溶解或乳化,只要其存在不干扰形成本发明的材料的方法即可。然而,为了避免干扰形成本发明的材料的方法,在优选实施方案中,本发明的方法是用步骤(a)中提供的水溶液进行的,该水溶液不含固体形式的未溶解物质,也不含有机溶剂。
[0098]
如果不使用掺杂金属和第二金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种有机的、至少部分水溶性的镍盐或多于一种的有机的、至少部分水溶性的镍盐的组合。在本发明中,如果盐的至少一部分在该方法中使用的条件下溶解在步骤(a)中提供的水溶液中,则认为盐至少部分是水溶性的。优选地,如果不使用掺杂金属且不使用第二金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多于一种有机镍盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0099]
在本发明的优选实施方案中,不使用掺杂金属和第二金属。
[0100]
在本发明的另一个优选实施方案中,使用掺杂金属,但不使用第二金属。
[0101]
如果使用掺杂金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机的、至少部分水溶性的镍盐与一种或多种掺杂金属的一种或多种有机的、至少部分水溶性的盐的组合。优选地,如果使用掺杂金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机镍盐与一种或多种掺杂金属的一种或多种有机盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0102]
如果使用第二金属,则在本发明方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机的、至少部分水溶性的镍盐与一种或多种第二金属的一种或多种有机的、至少部分水溶性的盐的组合。优选地,如果使用第二金属,则在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机镍盐与一种或多种第二金属的一种或多种有机盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0103]
如果使用掺杂金属和第二金属,在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机的、至少部分水溶性的镍盐与一种或多种掺杂金属的一种或多种有机的、至少部分水溶性的盐和一种或多种第二金属的一种或多种有机的、至少部分水溶性的盐的组合。优选地,如果使用掺杂金属和第二金属,在本发明的方法的步骤(a)中提供的溶液中的金属前体是一种或多种有机镍盐与一种或多种掺杂金属的一种或多种有机盐和一种或多种第二金属的一种或多种有机的、至少部分水溶性的盐的组合,其中所需包括在溶液中的量完全可溶于步骤(a)的水溶液中。
[0104]
在本发明的方法的步骤(a)中提供的溶液中,金属前体的优选有机阴离子是乙酸根、碳酸根、草酸根、柠檬酸根、丙二酸根、酒石酸根和戊二酸根。如果不需要避免氮,则硝酸根是步骤(a)中提供的溶液中金属前体的另一个优选阴离子。
[0105]
饱和的脂族二羧酸、三羧酸或多羧酸,单独或作为混合物的一部分,可以用作步骤
(a)中提供的水溶液的有机碳源,只要其支持本发明的材料的形成。在优选实施方案中,丙二酸、戊二酸、柠檬酸或其混合物用作本发明的方法的步骤(a)中提供的水溶液的有机碳源。在本发明的特别优选的实施方案中,柠檬酸用作本发明的方法的步骤(a)中提供的水溶液的有机碳源。
[0106]
在本发明方法的步骤(b)中,对在步骤(a)中提供的水溶液进行喷雾干燥或冷冻干燥。由此获得的产物在本发明的上下文中称为中间产物p。喷雾干燥和冷冻干燥的工艺参数可以在宽范围内变化,只要干燥过程不间断进行,并且中间产物p所表现出的水和有机溶剂的组合含量低于10重量%即可。在本发明的优选实施方案中,在本发明的方法的步骤(b)中对步骤(a)中提供的水溶液进行喷雾干燥。
[0107]
根据本发明的方法的步骤(c)的热处理是在规定的温度条件和惰性气体气氛(如氮气或空气)下进行的。用于此目的的多种合适的炉可商购获得。在优选实施方案中,热处理在惰性气体气氛(例如氮气)下进行。热处理过程中的加热速率应足够小,以使热量均匀分布,即通常小于15k/min,优选小于10k/min,特别优选小于5k/min。中间产物p在200℃-380℃的温度范围内进行热处理。在本发明的优选实施方案中,中间产物p在255℃-375℃的温度范围内进行热处理。在特别优选的实施方案中,中间产物p在300℃-350℃的温度范围内进行热处理。通常,对中间产物p进行1-4小时的热处理,但延长或缩短时间间隔的热处理也可以起作用。当确定热处理的持续时间时,不考虑加热和冷却间隔。在优选实施方案中,对所述中间产物p进行1-4小时的热处理。
[0108]
如上所述,根据本发明的材料表现出催化活性。因此,在另一方面,本发明进一步涉及本发明的材料作为催化剂的用途。
[0109]
根据本发明的材料例如可用作有机化合物的液相氢化的催化剂,所述有机化合物特别是不饱和化合物如烯烃、不饱和羧酸和不饱和醇、炔烃、醛和酮、酯和亚胺、硝基化合物和腈以及芳基化合物。此外,根据本发明的材料是用于羰基化合物的还原胺化的活性非常强的催化剂。因此,在另一方面,本发明进一步涉及本发明的材料作为用于有机化合物的氢化、羰基化合物的还原胺化和/或有机化合物的加氢甲酰化的催化剂的用途。
[0110]
根据本发明的材料还可以用作用氢气将一氧化碳、二氧化碳或其混合物转化为饱和及不饱和的烃或其混合物中的催化剂。因此,在另一方面,本发明还涉及本发明的材料作为用氢气将一氧化碳、二氧化碳或其混合物转化为甲烷的催化剂的用途。
[0111]
根据本发明的材料可以作为未改性形式的催化剂使用,或者可以通过本领域技术人员所公知的成型方法(例如压片、造粒、挤出、涂覆、3d打印)转化成催化剂体。
实施例
[0112]
实施例ni a、b-碳嵌入的ni纳米粒子(carbon embedded ni-nanoparticles)的制备
[0113]
通过在室温下连续搅拌,将14.4g柠檬酸(特纯(puriss),sigma aldrich)溶解于75ml去离子水中,制备碳嵌入的ni纳米粒子。在第二个烧杯中,在室温下连续搅拌下,将18.7g乙酸镍(ii)四水合物(ni(ch3coo)2*4h2o,sigma aldrich)溶解于75ml去离子水中。将乙酸镍溶液缓慢加入到柠檬酸溶液中,并在室温下再搅拌30分钟。用常规小型喷雾干燥机(b
ü
chi,mini spray dryer b-290)喷雾干燥所得溶液,所述常规小型喷雾干燥机具有220
℃的恒定入口温度、120℃的出口温度和20%的泵速。将获得的粉末分成质量相同的两部分进行最终的热处理。
[0114]
将第一个样品在氮气气氛下的管式炉中以180分钟升温(ramp)至300℃进行热处理,其中将温度再保持4小时,然后自然冷却。得到的催化剂粉末标记为nicat.1a。
[0115]
将第二个样品在氮气气氛下以类似的方式热处理。在180分钟内将样品加热至350℃,其中将温度保持4小时,然后自然冷却。得到的催化剂粉末标记为nicat.1b。
[0116]
使用在100kev下操作的配备有ccd照相机的校准的hitachi h-7500场透射电子显微镜,通过xrf(x射线荧光)和tgz分析,确定了这些材料具有以下特征:
[0117]
idd
p
ωdnicat.1a9.5nm0.5315nmnicat.1b11nm0.5812nm
[0118]
比较例
[0119]
为了比较,在传统的vulcan xc72r碳载体上,用初期湿浸渍法(incipient wetness impregnation)制备了镍含量为20%的高负载催化剂,并将其标记为nicat.ref。
[0120]
使用在100kev下操作的配备有ccd照相机的校准的hitachi h-7500场透射电子显微镜,通过xrf(x射线荧光)和tgz分析,确定了这些材料具有以下特征:
[0121]
idd
p
ωdnicat.ref67nm0.20n.d.*
[0122]
催化活性试验
[0123]
用200mg催化剂和5mmol底物在5ml甲醇中分批进行试验,以测定材料的催化活性和选择性。对于所有实验,将高压釜加热至所需的反应温度,并在50bar的恒定氢气压力下搅拌。过滤反应产物并通过gc-ms进行分析。
[0124]
i.n-亚苄基-苄胺的氢化
[0125][0126]
idcat.id持续时间h温度t℃反应物%产物%副产物%1nicat.1a20.00100.002.4088.98.702nicat.1b20.00100.001.7095.03.303nicat.ref20.00100.0064.023.612.4
[0127]
ii.巴豆酸甲酯氢化成丁酸甲酯
[0128][0129]
idcat.id持续时间h温度t℃反应物%产物%4nicat.1a20.0080.000.00100.005nicat.1b20.0080.000.00100.006nicat.ref20.0080.001.198.9
[0130]
iii.十二腈(dodecannitrile)的氢化
[0131][0132]
idcat.id持续时间h温度t℃反应物%产物%副产物%7nicat.1a20.0080.0065.528.85.68nicat.1b20.0080.00073.027.09nicat.ref20.0080.0072.520.17.4
[0133]
iv.乙酰基萘的氢化
[0134][0135]
idcat.id持续时间h温度t℃反应物%产物%副产物%10nicat.1a16.0080.000.00100.000.0011nicat.1b16.0080.000.00100.000.0012nicat.ref20.0080.000.00100.000.00
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。