1.本发明属于有机合成技术领域,具体涉及一种制备抗病毒药物替诺福韦艾拉酚胺富马酸盐的方法。
背景技术:
2.目前制备替诺福韦艾拉酚胺富马酸盐方法是由替诺福韦作为起始物料的,路线如下:
[0003][0004]
其中,单酯中间体a是由膦酸三苯酯与替诺福韦制备得到。
[0005]
由于替诺福韦艾拉酚胺富马酸盐的原料药注册申报不能使用替诺福韦作为替诺福韦艾拉酚胺富马酸盐的起始物料,合成路线必须往前延申。制备替诺福韦的常用方法,路线如下:
[0006][0007]
由上述合成路线可知,tf是经由中间体b水解得到的。研究发现当只水解掉一个乙基时,室温条件下就可以完成,而水解第二个乙基的反应速率非常慢,需要回流条件下才能在数小时完全水解掉。也就是说如果需要,可以控制反应条件选择性得到单酯中间体。
技术实现要素:
[0008]
针对上述技术问题,本发明提供一种替诺福韦艾拉酚胺富马酸盐的合成路线,该合成路线相比于现有合成路线步骤少,水解反应条件温和,氢溴酸用量减少一半,副反应少。
[0009]
本发明提供的技术方案如下:
[0010]
一种制备抗病毒药物替诺福韦艾拉酚胺富马酸盐新方法,包括以下步骤:
[0011]
(1)将腺嘌呤和(r)-碳酸丙烯酯溶于第一有机溶剂中进行反应,得到化合物i;
[0012]
化合物i为
[0013]
(2)将化合物i、叔丁醇镁溶于第二有机溶剂中,滴加膦酸酯s,进行保温反应,得到化合物ii;
[0014]
所述膦酸酯s为化合物ii为
[0015]
(3)将化合物ii加入氢溴酸中进行水解,经提纯后得到化合物iii;
[0016]
化合物iii为
[0017]
(4)将化合物iii与二氯亚砜反应,反应的产物溶于第三有机溶剂中得到溶液a;
[0018]
(5)将l-丙氨酸异丙酯溶于第三有机溶剂中,滴加溶液a进行反应,经提纯得到化合物iv;
[0019]
(6)将化合物iv溶于第四有机溶剂中,加入富马酸进行反应,得到富马酸丙酰酚替诺福韦。
[0020]
进一步,所述步骤(1)中,第一有机溶剂为dmf,反应采用加热回流,碱催化反应的方式进行,。
[0021]
进一步,所述步骤(2)中,第二有机溶剂为dmf,反应温度为70-80℃。
[0022]
进一步,所述步骤(2)中,化合物i、叔丁醇镁与膦酸酯s的摩尔用量比为1:0.6~1:1.1~1.5。
[0023]
进一步,所述步骤(3)中,反应温度为室温,反应时间为6-10小时。
[0024]
进一步,所述步骤(4)和步骤(5)中,第三有机溶剂为dcm。
[0025]
进一步,所述步骤(4)中,化合物iii与二氯亚砜的用量比为1g:1.0~10ml。
[0026]
进一步,所述步骤(4)中,反应温度为-30℃~-20℃。
[0027]
进一步,所述步骤(6)中,第四有机溶剂为丙酮。
[0028]
进一步,所述步骤(6)中,所述化合物iv与富马酸的摩尔用量比为1:0.5~1。
[0029]
本发明的有益效果如下:
[0030]
本发明通过反应物筛选和路线优化,合成膦酸混合酯作为中间体,合成路线步骤少,水解反应条件温和,氢溴酸用量节省一半。此外,由于强酸性水解反应温度由回流降到了室温,减少了一些副反应的发生,提高终产物得率。
附图说明
[0031]
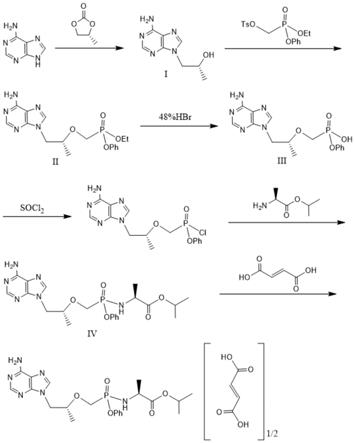
图1为本发明的合成路径图。
具体实施方式
[0032]
下面结合具体实施例对本发明进一步说明,本发明的内容完全不限于此。
[0033]
本发明提供的合成路线如图1所示,具体步骤如下:
[0034]
包括以下步骤:
[0035]
(1)将腺嘌呤和(r)-碳酸丙烯酯溶于第一有机溶剂中进行反应,得到化合物i;
[0036]
化合物i为
[0037]
(2)将化合物i、叔丁醇镁溶于第二有机溶剂中,滴加膦酸酯s,进行保温反应,得到化合物ii;
[0038]
所述膦酸酯s为化合物ii为
[0039]
(3)将化合物ii加入氢溴酸中进行水解,经提纯后得到化合物iii;
[0040]
化合物iii为
[0041]
(4)将化合物iii与二氯亚砜反应,反应的产物溶于第三有机溶剂中得到溶液a;
[0042]
(5)将l-丙氨酸异丙酯溶于第三有机溶剂中,滴加溶液a进行反应,经提纯得到化合物iv;
[0043]
(6)将化合物iv溶于第四有机溶剂中,加入富马酸进行反应,得到富马酸丙酰酚替诺福韦。
[0044]
进一步,所述步骤(1)中,第一有机溶剂为dmf,反应采用加热回流,碱催化反应的方式进行。
[0045]
进一步,所述步骤(2)中,第二有机溶剂为dmf,反应温度为70-80℃。
[0046]
进一步,所述步骤(2)中,化合物i、叔丁醇镁与膦酸酯s的摩尔用量比为:1:0.6~1:1.1~1.5。
[0047]
进一步,所述步骤(3)中,反应温度为室温,反应时间为6-10小时。
[0048]
进一步,所述步骤(4)和步骤(5)中,第三有机溶剂为dcm。
[0049]
进一步,所述步骤(4)中,化合物iii与二氯亚砜的用量比为1g:1.0~10ml。
[0050]
进一步,所述步骤(4)中,反应温度为-30℃~-20℃。
[0051]
进一步,所述步骤(6)中,第四有机溶剂为丙酮。
[0052]
进一步,所述步骤(6)中,所述化合物iv与富马酸的摩尔用量比为1:0.5~1。
[0053]
实施例
[0054]
合成步骤如下:
[0055]
(1)化合物i的合成
[0056]
于反应瓶中加入100g腺嘌呤和0.9g氢氧化钠,120ml dmf,开启搅拌。加入91g(r)-碳酸丙烯酯后,加热回流6小时。降温至70℃,滴加醋酸的水溶液,调ph=6~7。再加入150ml乙醇。加完,降温至0~10℃析晶。过滤,滤饼用乙醇淋洗,干燥后得到115g化合物i,收率80%。
[0057]
(2)化合物ii的合成
[0058]
于反应瓶中搅拌下分别加入200ml dmf、100g化合物i和68g叔丁醇镁,在70-80℃,搅拌1小时。然后开始滴加230g膦酸酯s滴加结束,保温反应3小时。将反应液降温至30
±
5℃后,滴加600g冰醋酸。减压浓缩至无液体流出。加入800ml二氯甲烷,和150ml水。水相再用150ml二氯甲烷萃取2次,合并有机相,减压浓缩至无溶剂流出,得到化合物ii的粗品,直接用于下一步。
[0059]
(3)化合物iii的合成
[0060]
向上步化合物ii粗品中加入300ml 48wt%氢溴酸,室温下搅拌反应8小时。加入300ml水,用400ml二氯甲烷萃取。水相用浓氨水,调ph至2.5~3。过滤,滤饼用水淋洗,干燥得185.8g化合物iii,两步收率85%。
[0061]
(4)化合物iv的合成
[0062]
于反应瓶中分别加入15g化合物iii和20ml二氯亚砜,加热回流过夜。减压除去过量的二氯亚砜,残留物溶于dcm得到溶于a。
[0063]
将13g l-丙氨酸异丙酯溶于100ml dcm,降温至-30℃。然后开始滴加溶液a,滴加过程控温低于-20℃。滴毕,继续搅拌2小时。反应液分别用稀盐酸、水、饱和碳酸氢钠水溶液和食盐水洗涤,无水硫酸镁干燥,过滤,减压浓缩得到化合物粗品。粗品经重结晶,脱色得到15g化合物iv,收率75%。
[0064]
(5)富马酸丙酰酚替诺福韦的合成
[0065]
将10g化合物iv溶于100ml丙酮中,搅拌下加入1.2g富马酸,加热回流2小时。降至室温析晶,过滤,干燥得到10g产品,收率90%。
[0066]
以上所述,仅为本发明较佳的具体实施方式,但本发明保护的范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内所做的任何修改,等同替换和
改进等,均应包含在发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。