作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物及制备方法和用途
技术领域
[0001]
本发明涉及化工医药化合物及制备方法和用途,特别涉及作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物及制备方法和用途。
背景技术:
[0002]
jak激酶(janus kinase)家族在哺乳动物体内有四种亚型:jak-1、jak-2、jak-3和tyk-2,在细胞素依赖性调节与生长和免疫应答相关的细胞的功能方面起一定作用。jak激酶和其下游的效应器、信号转导及转录激活蛋白(signal transducers and activators of transcription proteins,stats)形成了重要的细胞因子信号传导途径——jak-stat通路(science,1994,264:1415-1421)。该通路可由多种细胞因子、生长因子以及受体激活,如白介素(il)类、干扰素(ifn)类、促红细胞生成素(epo)、粒细胞和巨噬细胞集落刺激因子(gmcsf)、促生长素(gh)、催乳素(prl)、促血小板生成素(tpo)、血小板衍生因子(pdgf)以及表皮细胞生长因子(egf)等,而且不同受体可激活不同亚型的jak激酶,参与细胞增殖、分化、凋亡、血管生成以及免疫调节等过程(world j gastroenterol,2007,13:6478-6491)。
[0003]
jak激酶家族中jak-1、jak-2和tyk-2在人体各组织细胞中均有表达,而jak-3主要表达于各造血组织细胞中,如骨髓细胞、胸腺细胞、nk细胞及活化的b淋巴细胞、t淋巴细胞中,jak-3通过与il-2、il-4、il-7、il-9、il-15和il-21等i型细胞因子受体复合物中的γ链(γc)相结合,调节细胞信号传导。当jak-3缺陷或γc突变时,可导致重症联合免疫缺陷(severe combined immunodeficiency,scid)(blood,1996,88:817-823),表现为t细胞和自然杀伤细胞(natural killer cell,nk)细胞减少、b细胞功能丧失等免疫限制的症状(chin j new drug,2015,24:39-45)。适量的细胞素在免疫应答中起重要作用,但当其过度产生则会诱发许多自身免疫疾病,如牛皮癣、类风湿性关节炎、炎性肠疾病、斯耶格伦氏综合征、白塞氏病、多发性硬化、系统性红斑狼疮等等(journal of allergy and clinical tmmurmlogy,2011,127:701-721;cytoki ne&growth factor reviews,2008,19:41-52;invest ophthalmol vis sci,2008,49:3058-3064;ann rheum dis,2010,69:1325-1328)。综上所述,jak激酶抑制剂是各种自身免疫疾病的潜在治疗药物,jak-3是公认的jak-stat通路中较为安全有效的抗自身免疫靶点。
技术实现要素:
[0004]
发明目的:本发明目的是提供作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物或其可药用的盐。另一目的是提供作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物或其可药用的盐的制备方法。最后一个目的是提供所述作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物或其可药用的盐的用途。
[0005]
本发明研究的重点是通过抑制atp与jak激酶上atp结合位点的结合,阻断atp的水解,干扰jaks磷酸化,从而阻止jaks的活化,切断其向stats传递信号,导致其无法调控细胞
7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺;(e)-n-(4-((4-甲基-2-((1-甲基-1h-吡唑-4-基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丁-2-烯酰胺;n-(4-((4-甲基-2-((1-甲基-1h-吡唑-4-基)氨基)-5,6-二氢-7h-吡咯并[2,3-d])嘧啶-7-基)甲基)苯基)氰胺;n-(4-((2-((1-(2-甲氧基乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺;n-(4-((2-((1-(2-甲氧基乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺;(e)-n-(4-((2-((1-(2-甲氧基乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丁-2-烯酰胺;n-(4-((2-((1-(2-甲氧基乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)氰胺;n-(4-((2-((1-(2-羟乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺;n-(4-((2-((1-(2-羟乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺;(e)-n-(4-((2-((1-(2-羟乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丁-2-烯酰胺;n-(4-((2-((1-(2-羟乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)氰胺;n-(4-((2-((1-(2-(二甲氨基)乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺;n-(4-((2-((1-(2-(二甲氨基)乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺;(e)-n-(4-((2-((1-(2-(二甲氨基)乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丁-2-烯酰胺;n-(4-((2-((1-(2-(二甲氨基)-2-氧乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺;n-(4-((2-((1-(2-(二甲氨基)-2-氧乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺;(e)-n-(4-((2-((1-(2-(二甲氨基)-2-氧乙基)-1h-吡唑-4-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丁-2-烯酰胺;2-(4-((7-(4-氰氨苄基)-4-甲基-6,7-二氢-5h-吡咯并[2,3-d]嘧啶-2-基)氨基)-1h-吡唑-1-基)-n,n-二甲基乙酰胺;n-(4-((4-甲基-2-((4-吗啉苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺;n-(4-((4-甲基-2-((4-吗啉苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺;(e)-n-(3-((4-甲基-2-((4-吗啉苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丁-2-烯酰胺;n-(4-((4-甲基-2-((4-吗啉苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)氰胺;n-(4-((4-甲基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺;n-(4-((4-甲基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺;(e)-n-(4-((4-甲基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丁-2-烯酰胺;n-(3-((4-甲基-2-((4-(4-甲基哌嗪-1-基)苯基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)氰胺;n-(4-((2-((1-羟基-1,3-二氢苯并[c][1,2]恶硼唑-6-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺;n-(4-((2-((1-羟基-1,3-二氢苯并[c][1,2]恶硼唑-6-基)氨基)-4-甲基-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)环丙基甲酰胺。
[0021]
一种药物组合物,其含有治疗有效量的一种或多种如权利要求1-6中任一项作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物或其可药用的盐,及药学上可接受的载体或辅料。
[0022]
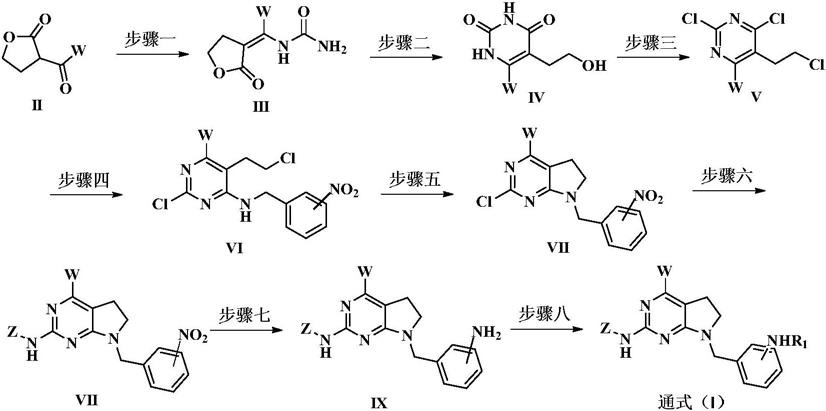
所述作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物或其可药用的盐,的制备方法,包括如下步骤:
[0023][0024]
其中,
[0025]
步骤一:化合物ii与尿素在乙醇中回流得到化合物iii;
[0026]
步骤二:化合物iii在碱性乙醇钠的作用下回流得到化合物iv;
[0027]
步骤三:化合物iv在三氯氧磷及三乙胺的条件下转化为v;
[0028]
步骤四;化合物v与取代苄胺反应得到化合物vi;
[0029]
步骤五:化合物vi在氢化钠的作用下得到化合物vii;
[0030]
步骤六:化合物vii与取代胺反应得到化合物viii;
[0031]
步骤七:化合物viii还原反应得到化合物ix;
[0032]
步骤八:化合物ix与r1取代试剂进行酰胺反应得到本发明化合物。
[0033]
所述的作为jak激酶抑制剂的吡咯并[2,3-d]嘧啶类衍生物或其可药用的盐,在制备治疗自身免疫疾病、斯耶格伦氏综合征、白塞氏病、多发性硬化、系统性红斑狼疮的药物中的用途。所述自身免疫疾病为牛皮癣、类风湿性关节炎或炎性肠疾病。
[0034]
本发明开发的高活性和选择性的jak抑制剂或其可药用的盐,竞争性抑制atp与jak激酶上atp结合位点的结合,阻断atp的水解,干扰jaks磷酸化,从而阻止jaks的活化,切断其向stats传递信号,导致其无法调控细胞核内基因的表达,达到阻断jak-stat信号通路的目的。吡咯并[2,3-d]嘧啶类化合物,作为预防、治疗或改善包括例如炎性疾病和自身免疫疾病(例如,类风湿性关节炎、银屑病、炎性肠炎疾病、系统性红斑狼疮、牛皮癣、斯耶格伦氏综合征、白塞氏病、多发性硬化等等)以及癌症(例如,巨大淋巴结增生症、淋巴瘤、白血病多发性骨髓瘤或骨髓增生性疾病等等)等等的jak相关性疾病的药物,还涉及含有它的药物组合、制备方法及用途。
[0035]
有益效果:本发明的化合物具有优异的jak-3抑制作用,其用作预防、治疗或改善自身免疫疾病(例如,牛皮癣、类风湿性关节炎、炎性肠疾病(例如,克罗恩病、溃疡性结肠炎
等等)、斯耶格伦氏综合征、白塞氏病、多发性硬化、系统性红斑狼疮等等)的药物,等等,实施例15、53、64、67在10μm浓度下表现出高活性的jak-3抑制作用,本发明合成路线简单,实施性强。
具体实施方式
[0036]
以下实施例中,“室温”是指大约10℃至大约35℃。混合溶剂表示的比例是体混合比例,除非另作说明,否则%是指wt%。
[0037]
在砫胶柱色谱中,碱性砫胶是指使用氨基丙基硅烷结合的硅胶。在高效液相色谱(hplc)中,c18是指使用十八烷基结合的硅胶。洗脱溶剂的比例是体积混合比例,除非另作说明。
[0038]
在下面实施例和实验实施例中,使用下列缩写。
[0039]
thf:四氢呋喃,
[0040]
dcm:二氯甲烷,
[0041]
dmso:二甲基亚砜,
[0042]
diea:n,n-二异丙基乙胺,
[0043]
koac:醋酸钾,
[0044]
et3n:三乙胺,
[0045]
m:摩尔浓度。
[0046]
利用fourier变换类型nmr,测定1h-nmr(质子核磁共振波谱)。对于分析,使用acd/specmanager等。不描述活性氢(例如羟基、氨基等等)的峰。
[0047]
利用lc/ms(液相色谱质谱仪)测定ms(质谱)。作为电离法,使用esi(电喷射离子化)方法等。数据表示那些实测值。通常,观察分子离子峰。在盐的情况下,通常观察到游离形式的分子离子峰或碎片离子峰。
[0048]
以下为部分y取代基的结构式及制备方法:
[0049]
实施例1
[0050]
n-甲基-4-氨基吡唑
[0051][0052]
将4-硝基吡唑(1.41g),碳酸钾(2.5g),碘甲烷(1.9g)和20ml乙腈,回流反应12小时。减压旋干,加入50ml水,分别用50ml乙酸乙酯萃取3次,合并有机层,饱和食盐水洗涤,有机相加入无水na2so4干燥,减压干燥,得1-甲基-4-硝基吡唑(1.5g)。
[0053]
将1-甲基-4-硝基吡唑(1g),pd/c(0.1g)和20ml乙醇,氢气球置换空气,室温反应约24小时。硅藻土减压抽滤,滤液减压干燥,得标题产物(0.75g)。ms(esi):[m h]
98.0m/z。
[0054]
实施例2
[0055]
1-(2-甲氧基乙基)-1h-吡唑-4-胺
[0056][0057]
利用与参考实施例1一样的方法,由4-硝基吡唑、2-溴甲基乙基醚、pd/c获得标题化合物。ms(esi):[m h]
142.0m/z。
[0058]
实施例3
[0059]
2-(4-氨基-1h-吡唑-1-基)乙醇
[0060][0061]
利用与参考实施例1一样的方法,由4-硝基吡唑、2-溴乙醇、pd/c获得标题化合物。ms(esi):[m h]
128.0m/z。
[0062]
实施例4
[0063]
n,n-二甲基-2-(4-氨基-1h-吡唑1-基)乙胺
[0064][0065]
利用与参考实施例1一样的方法,由4-硝基吡唑、n,n-二甲基-2-溴-乙胺、pd/c获得标题化合物。ms(esi):[m h]
154.1m/z。
[0066]
实施例5
[0067]
n-甲基-2-(4-氨基-1h-吡唑1-基)乙酰胺
[0068][0069]
利用与参考实施例1一样的方法,由4-硝基吡唑、n-甲基-2-溴-乙酰胺、pd/c获得标题化合物。ms(esi):[m h]
154.0m/z。
[0070]
实施例6
[0071]
n,n-二甲基-2-(4-氨基-1h-吡唑1-基)乙酰胺
[0072][0073]
利用与参考实施例1一样的方法,由4-硝基吡唑、n,n-二甲基-2-溴-乙酰胺、pd/c获得标题化合物。ms(esi):[m h]
168.1m/z。
[0074]
实施例7
[0075]
4-(4-吗啉基)苯胺
[0076][0077]
将4-氟硝基苯(1g),碳酸钾(1.1g)和5ml dmso,于室温搅拌反应30分钟,滴加吗啡啉(0.6g),加热120℃搅拌反应2小时,混合物倒入醇和水混合液(1∶1)20ml中,过滤得黄色沉淀4-(4-硝基苯基)吗啉1.4g。
[0078]
将4-(4-硝基苯基)吗啉(1g),pd/c(0.1g)和20ml乙醇,于氢气球置换空气,室温反应约24小时。硅藻土减压抽滤,滤液减压干燥,得标题产物(0.85g)。ms(esi):[m h]
179.1m/z。
[0079]
实施例8
[0080]
4-(4-甲基哌嗪-1-基)苯胺
[0081][0082]
利用与参考实施例7一样的方法,由4-氟硝基苯、n-甲基哌嗪、pd/c获得标题化合物。ms(esi):[m h]
191.1m/z。
[0083]
实施例9
[0084]
6-氨基苯并[c][1,2]氧杂硼杂环戊-1(3h)-醇
[0085][0086]
将2-甲酰基苯基硼酸(1g)分布于thf中,冷却至-30℃,加入氢化钠,-30℃搅拌反应10分钟,室温反应30分钟。减压旋干,加入20ml水,分别用20ml乙酸乙酯萃取3次,合并有机层,饱和食盐水洗涤,有机相加入无水na2so4干燥,减压干燥,得苯并[c][1,2]氧杂硼杂环戊-1(3h)-醇(0.76g)。
[0087]
将苯并[c][1,2]氧杂硼杂环戊-1(3h)-醇(0.5g)-40℃条件下分批加入发烟硝酸0.3ml,搅拌反应30分钟,移至室温,混合物倒入冰水20ml中,过滤得黄色沉淀6-硝基苯并[c][1,2]氧杂硼杂环戊-1(3h)-醇(0.5g)。
[0088]
将6-硝基苯并[c][1,2]氧杂硼杂环戊-1(3h)-醇(0.5g),pd/c(0.05g)和10ml甲醇,于氢气球置换空气,室温反应约24小时。硅藻土减压抽滤,滤液减压干燥,得标题产物(0.4g)。ms(esi):[m h]
149.9m/z。
[0089]
以下为本技术部分化合物的制备方法:
[0090]
实施例10
[0091]
n-(4-((4-甲基-2-((1-甲基-1h-吡唑-4-基)氨基)-5,6-二氢-7h-吡咯并[2,3-d]嘧啶-7-基)甲基)苯基)丙烯酰胺
吡咯并[2,3-d]嘧啶-2-胺(0.1g),diea(0.03g),10ml干燥dcm中,滴加1ml dcm稀释的丙烯酰氯,搅拌反应30分钟,抽滤可得到标题化合物。1h nmr(300hz,cd3od)δ7.80(s,1h),7.63(s,1h),7.59(s,1h),7.46(d,j=7.9hz,1h),7.17(t,j=7.8hz,1h),6.93(d,j=4.8hz,1h),6.36(dd,j=16.9,10.1hz,1h),6.16(d,j=16.8hz,1h),5.58(d,j=11.5hz,1h),4.54(s,2h),3.73(s,3h),3.65-3.55(m,2h),2.79(t,j=7.3hz,2h),2.05(s,3h).ms(esi):[m h]
390.2033m/z。
[0108]
实施例11至56
[0109]
在实施例11至17,利用与实施例10一样的方法。
[0110]
其中,实例例18至25,在步骤c)中3-硝基苄胺(1g)对应换为4-硝基苄胺(1g),其他条件不变。
[0111]
根据实施例结构式,其步骤e)对应不同的氨基烷基化的取代基,其具体是y取代基(上述有说明)。
[0112]
其中实施例26至41,对应步骤g)中0℃滴加1ml干燥dcm稀释的丙烯酰氯换成0℃滴加1ml干燥dcm稀释的环丙基甲酰氯,其他条件不变。
[0113]
其中实施例42至56,对应步骤g)中0℃滴加1ml干燥dcm稀释的丙烯酰氯换成0℃滴加1ml干燥dcm稀释的丁烯酰氯,其他条件不变。以上摩尔当量比与实施例10对应的反应当量相同。
[0114]
继而可获得以下标题化合物,见表1。该表中的ms是指实测值。
[0115]
表1部分化合物结构及核磁数据
[0116]
[0117]
[0118]
[0119]
[0120]
[0121]
[0122]
[0123]
[0124]
[0125]
[0126]
[0127][0128]
实施例57
[0129]
n-(3-((4-甲基-2-((1-甲基-1h-吡唑-4-基)氨基)-5,6-二氢-7h-吡咯并[2,3-d])嘧啶-7-基)甲基)苯基)氰胺
[0130]
实施步骤a)、b)、c)、d)、e)、f)利用和实施例一一样的方法,可获得7-(3-氨基苄基)-4-甲基-n-(1-甲基-1h-吡唑-4-基)-6,7-二氢-5h-吡咯并[2,3-d]嘧啶-2-胺。
[0131]
h)n-(3-((4-甲基-2-((1-甲基-1h-吡唑-4-基)氨基)-5,6-二氢-7h-吡咯并[2,3-d])嘧啶-7-基)甲基)苯基)氰胺
[0132]
向7-(3-氨基苄基)-4-甲基-n-(1-甲基-1h-吡唑-4-基)-6,7-二氢-5h-吡咯并[2,3-d]嘧啶-2-胺,koac和10ml干燥甲醇中,0℃搅拌下缓慢滴加甲醇溶解稀释的溴氰,后转至室温反应。反应结束,加入dcm,饱和na2co3水溶液洗涤两次,有机相拌样,经硅胶柱色谱分离可得标题化合物。
[0133]1h nmr(300mhz,chloroform-d)δ7.71-7.37(m,2h),7.31(t,j=7.7hz,1h),7.04(d,j=7.6hz,1h),6.92(s,2h),4.60(s,2h),3.69(t,j=7.5hz,2h),3.61(s,3h),2.87(t,j=6.8hz,2h),2.15(s,3h).
[0134]
ms(esi):[m h]
361.2m/z。
[0135]
实施例58至64
[0136]
在实施例,利用与实施例57一样的方法。
[0137]
其中,实例例67至72,在步骤c)中3-硝基苄胺(1g)对应换为4-硝基苄胺(1g),其他条件不变。
[0138]
根据实施例结构式,其步骤e)对应不同的氨基烷基化的取代基,其具体是y取代基(上述有说明)。
[0139]
以上摩尔当量比与实施例10对应的反应当量相同。
[0140]
继而可获得以下标题化合物,见表2。该表中的ms是指实测值。
[0141]
表2部分化合物结构及核磁数据
[0142]
[0143]
[0144][0145]
[0146]
实施例70:实验实施例(jak-3激酶抑制试验)
[0147]
实验目的:
[0148]
评价化合物对jak-3激酶的抑制作用。
[0149]
实验原理:
[0150]
基于荧光共振能量转移技术(fret)偶联蛋白水解酶对特定磷酸化与非磷酸化多肽底物不同的蛋白水解作用。多肽底物两端分别标记为fret能量供体香豆素和能量受体荧光素,当供、受体距离较近时,可激发供体可以发生能量转移。
[0151]
激酶反应(kinase reaction)中,jak-3可以将atp中的γ-磷酸转移到多肽底物的单个酪氨酸残基上,如果体系中存在jak-3抑制剂,则atp上的γ-磷酸基团不会被转移到底物多肽上,磷酸化反应不能发生。基于此原理设计了激酶抑制剂的评价实验,底物多肽设计有激酶磷酸化位点,同时也是蛋白酶切位点,两端分别接2种荧光基团,分别为供体和受体。如果反应体系中激酶活性保持,γ-磷酸基团则被转移到底物的酶切位点,从而不会被蛋白酶切割而分离成两段,在特定波长激光激发下,一段荧光的能量会被转移到另一端的荧光基团而发射能量。否则,激酶活性被抑制后,磷酸基团不能被转移,底物酶切位点会被体系中的酶切割,底物分离成两段,则不会发生荧光的能量转移。基于此评价激酶的活性。
[0152]
实验步骤:
[0153]
本实验选择10μl激酶反应体系,jak3抑制剂筛选中,每体系含jak3激酶0.012ng/μl,atp 1.43μm。tk substrate-biotin底物为1μm,化合物初筛浓度10μm。
[0154]
(1)激酶反应缓冲液的准备
[0155]
将1ml 5
×
kinase buffer加入4ml双蒸水中稀释至1
×
,加入5μl 1m dtt及25μl 1m mgcl2(jak1反应缓冲液加5μl 1m mncl2)命名为kinase buffer室温储存。
[0156]
(2)待筛选化合物配置
[0157]
样品化合物按质量,溶于dmso,配成浓度为100mm的母液,用激酶反应缓冲液稀释和配置化合物,化合物的反应终浓度为10μm。
[0158]
(3)反应体系
[0159]
本实验载板为黑色384孔微量板,反应体系为10μl。
[0160]
荧光检测
[0161]
在330nm激发下,检测665nm和620nm的发射强度。
[0162]
酶标仪自动计算ratio=665/620
×
10000
[0163]
(5)数据分析
[0164]
按下式计算每个样品的抑制率
[0165]
抑制率=(ratio max-ratio sample)/(ratio max-ratio neg)
×
100%
[0166]
表三实验结果
[0167][0168]
结果显示,上述12个化合物在10μm浓度下对jak3激酶表现出一定的抑制作用,部分实施例化合物如实施例15、53、64、67表现出高于阳性药托法替尼及瑞莱昔替尼的抑制活性。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。