一种中间体
α-氯代乙酰基-γ-丁内酯的合成方法及硫噻唑的合成方法
技术领域
1.本发明涉及硫噻唑合成技术领域,具体涉及一种中间体α-氯代乙酰基-γ-丁内酯的合成方法及硫噻唑的合成方法。
背景技术:
2.4-甲基-5-(β羟乙基)-噻唑(简称硫噻唑),是一种常用香料。其根据生产条件不同包括肉香型、豆香型及奶香型产品,主要用于肉类,调味品和海产品的加香。不仅如此,硫噻唑是生产b类维生素,消炎和活血类药物的主要原料。硫噻唑作为一种重要的工业原料,国内国际市场潜力巨大。现有合成硫噻唑的工艺方法较多,目前主要有以下几种:
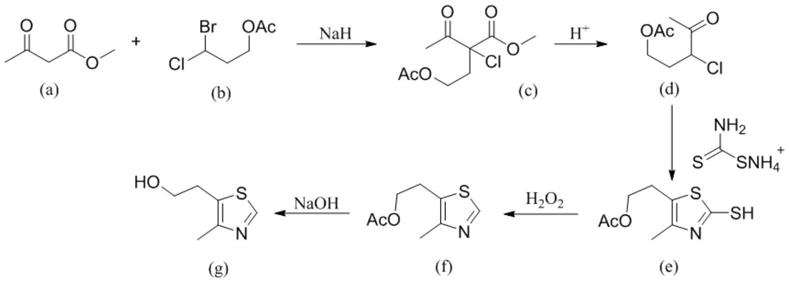
3.(1)以乙酰乙酸甲酯(a)和3-溴-3-氯丙基乙酸酯(b)为原料,在强碱性条件下反应得到4-乙酰氧基-2-乙酰基-2-氯丁酸甲酯(c),中间体(c)水解得到3-氯-3-乙酰丙醇乙酸酯(d),(d)与二硫代氨基甲酸铵在酸性条件下制备得到中间体(e),中间体e经氧化得到4-甲基-5-(β-乙酰氧乙基)-噻唑,最终在碱性条件下转化得到目标产物(g)(如式1所示)。该工艺路线步骤较多,原料3-溴-3-氯丙基乙酸酯(b)价格昂贵,来源受限,中间体(c)水解反应伴有较多副反应发生,反应效率降低,后续纯化过程复杂。且该路线中应用的氢化钠化学反应活性很高,不易储存,还具有一定的危险性。
[0004][0005]
式1.乙酰乙酸乙酯为原料合成硫噻唑
[0006]
(2)以2,3-二氯-2-甲基-4,5-二氢呋喃(h)或相应的二溴化合物为原料,与硫代甲酰胺反应得到目标产物(g)(如式2所示)。此方法虽然工艺流程简短,但其原料2,3-二氯-2-甲基-4,5-二氢呋喃(h)来源困难,现今国内仍未开发出可商业化市售产品,因此该路线不适合工业化生产。
[0007][0008]
式2.2,3-二氯-2-甲基-4,5-二氢呋喃为原料合成硫噻唑
[0009]
(3)以乙酸乙酯(m)和γ-丁内酯(n)为原料,金属钠为缩合剂制备α-乙酰基-γ-丁
内酯(i),后经氯化得到α-氯代乙酰基-γ-丁内酯(j),之后水解得到关键中间体3-氯-4-氧-1-戊醇(k),再与硫代甲酰胺类衍生物发生反应关环得到4-甲基-5-(β羟乙基)-噻唑衍生物(l),最后得到目标产物(g)(如式3所示)。
[0010][0011]
式3.γ-丁内酯为原料合成硫噻唑
[0012]
国内目前主要以该工艺路线工业生产硫噻唑。该技术路线中α-乙酰基-γ-丁内酯(i)为原料经氯化得到α-氯代乙酰基-γ-丁内酯(j)是关键步骤。α-乙酰基-γ-丁内酯(i)到目标产物可采用一锅法实现。该方法产品质量高、生产成本低,可实现工业化。国内主要以cl2为氯化剂,将α-乙酰基-γ-丁内酯(i)氯化为关键中间体α-氯代乙酰基-γ-丁内酯(j)。该路线虽步骤较少,工艺简单,但生产过程中涉及大量的酸和碱原料,大大增加了三废处理负担,增加生产成本,不符合绿色化学要求。且cl2是剧毒化工原料,在化工生产中具有一定安全危险性,使用和生产管理复杂。并且该路线对设备要求很高,增加了安全维护及设备运行的成本。
技术实现要素:
[0013]
为了解决采用cl2为氯化剂将α-乙酰基-γ-丁内酯(i)氯化为关键中间体α-氯代乙酰基-γ-丁内酯(j),生产过程中涉及大量的酸和碱原料,大大增加了三废处理负担,增加生产成本,不符合绿色化学要求;且cl2是剧毒化工原料,在化工生产中具有一定安全危险性的技术问题。
[0014]
本发明提供了一种中间体α-氯代乙酰基-γ-丁内酯的合成方法及硫噻唑的合成方法,以α-乙酰基-γ-丁内酯为原料,采用廉价、安全的三氯异氰尿酸为氯化试剂,通过氯化反应合成得到α-氯代乙酰基-γ-丁内酯,所制备的α-氯代乙酰基-γ-丁内酯应用于制备硫噻唑。本发明使用三氯异氰尿酸作为氯化剂,与传统氯化工艺采用氯气进行氯化过程相比,氯化反应平稳,选择性好,使用方便,安全性高。该方法氯化副产物为氰尿酸,可回收循环使用,相比氯气,无需处理反应后产生大量的盐酸和次氯酸,减轻三废处理负担,具有工业化生产的前景。
[0015]
本发明涉及三氯异氰尿酸在硫噻唑合成中应用。以α-乙酰基-γ-丁内酯为原料,在三氯异氰尿酸作用下,经氯化得到α-氯代乙酰基-γ-丁内酯,之后水解得到关键中间体3-氯-4-氧-1-戊醇,后与硫代甲酰胺发生反应关环得到目标产物。合成路线反应,参见式4:
[0016]
[0017]
式4
[0018]
本发明提供了一种中间体α-氯代乙酰基-γ-丁内酯的合成方法,以α-乙酰基-γ-丁内酯为原料,加入三氯异氰尿酸,经氯化得到α-氯代乙酰基-γ-丁内酯,反应式参见式5:
[0019][0020]
式5.tcca氯化α-乙酰基-γ-丁内酯反应
[0021]
该合成方法具体包括以下步骤:
[0022]
向反应烧瓶中依次加入一定比例量α-乙酰基-γ-丁内酯(也可以添加反应溶剂),开启搅拌,在10℃条件下,慢慢向上述反应混合溶液中加入所需量的三氯异氰尿酸。加料完毕后,加热反应混合液至设定反应温度,反应温度为10~80℃,优选25~50℃,持续搅拌使反应体系均匀,反应时间为0.5~12h,优选2~4h。反应完成后,采用气相色谱检测α-乙酰基-γ-丁内酯是否反应完全。冷却反应混合液,过滤分离反应生成的副产物氰尿酸,滤液为无色或淡黄色液体,即α-氯代乙酰基-γ-丁内酯粗产品。如果反应体系添加有反应溶剂,则过滤分离反应生成的副产物氰尿酸后的滤液经蒸馏脱除反应溶剂,得无色或淡黄色液体,即α-氯代乙酰基-γ-丁内酯粗产品。气相色谱检测α-氯代乙酰基-γ-丁内酯粗产品,α-氯代乙酰基-γ-丁内酯粗产品的含量(即纯度吗)在95%以上,可直接应用于合成硫噻唑。
[0023]
上述tcca氯化α-乙酰基-γ-丁内酯反应中:
[0024]
(1)反应可在有溶剂或无溶剂条件下进行。α-乙酰基-γ-丁内酯常温下为液态,优选无溶剂反应体系。当反应在有机溶剂条件下进行时,有机溶剂可为二氯甲烷、1,2-二氯乙烷、乙醚、thf和乙腈等,优选二氯甲烷,1,2-二氯乙烷。α-乙酰基-γ-丁内酯与有机溶剂质量比为1:1~10,优选1:3~6;
[0025]
(2)α-乙酰基-γ-丁内酯原料与氯化剂三氯异氰尿酸之间摩尔比例为1:0.33~3.0,优选1:0.35~0.8。
[0026]
本发明还提供了一种硫噻唑的合成方法,包括以下步骤:
[0027]
步骤1:中间体α-氯代乙酰基-γ-丁内酯的合成:
[0028]
采用上述中间体α-氯代乙酰基-γ-丁内酯的合成方法合成α-氯代乙酰基-γ-丁内酯。
[0029]
步骤2:中间体3-氯-4-氧-1-戊醇的合成:
[0030]
α-氯代乙酰基-γ-丁内酯水解开环合成3-氯-4-氧-1-戊醇的合成,反应方程式参见式6:
[0031][0032]
式6.3-氯-4-氧-1-戊醇的合成
[0033]
向反应烧瓶中加入一定量的α-氯代乙酰基-γ-丁内酯,向其中加入一定浓度的稀酸,加热回流反应混合液,反应温度为20-160℃,优选100-140℃,反应时间1-16小时,优选
3-9小时。反应完成后,气相色谱检测α-氯代乙酰基-γ-丁内酯是否反应完全。冷却反应液至室温,用质量分数5%的naoh水溶液调节反应ph至7-8,静置,分出有机相,水相用二氯甲烷萃取,将萃取相和有机相混合,混合液经干燥后,旋转蒸发脱除脱去溶剂,再减压蒸馏,得到目标产物中间体3-氯-4-氧-1-戊醇,气相色谱检测目标产物中间体3-氯-4-氧-1-戊醇粗产物,目标产物中间体3-氯-4-氧-1-戊醇的含量高于96.5%。
[0034]
上述3-氯-4-氧-1-戊醇的合成反应中:
[0035]
(1)该反应中稀酸与原料α-氯代乙酰基-γ-丁内酯的摩尔比例为1:1-20,优选1:5-15;
[0036]
(2)所述稀酸为无机酸,优选盐酸,硫酸。若选用盐酸,则酸的浓度为2%-30%,优选5%-20%。若选用硫酸,则酸的浓度为0.5%-98%,优选3%-15%。
[0037]
步骤3:4-甲基-5-(β羟乙基)-噻唑的合成:
[0038]
3-氯-4-氧-1-戊醇与硫代甲酰胺反应合成硫噻唑,反应方程式参见式7:
[0039][0040]
式7.硫噻唑的合成
[0041]
向反应烧瓶中加入一定量的3-氯-4-氧-1-戊醇,在低温条件(0-20℃之间,优选0-5℃)下向反应体系中加入相应量的稀酸,保持搅拌,向其中加入所需量的硫代甲酰胺。加入完毕后加热反应至设定的反应温度,反应温度为0-140℃,优选90-120℃,反应时间1-12小时,优选1.5-6小时。反应完成后,气相色谱检测3-氯-4-氧-1-戊醇是否反应完全,计算收率。冷却反应液至室温,加入naoh调节ph至相应值7-13,优选8-10,静置分层,分出有机相,水相用二氯甲烷萃取,将萃取相和有机相混合,混合液经干燥后,旋转蒸发脱除脱去溶剂,再减压蒸馏,得到目标产物硫噻唑。
[0042]
上述硫噻唑的合成反应中:
[0043]
(1)所述原料3-氯-4-氧-1-戊醇与酸的摩尔比例为1:0.01-0.6,优选1:0.01-0.1;
[0044]
(2)所述原料3-氯-4-氧-1-戊醇与硫代甲酰胺的摩尔比例为1:1-5,优选1:1.2-2;
[0045]
(3)所述反应酸为盐酸,硫酸,优选浓度为0.5-5%的硫酸。
[0046]
本发明还提供了一种三氯异氰尿酸作为氯化剂在制备中间体α-氯代乙酰基-γ-丁内酯或硫噻唑中的应用。
[0047]
与现有技术相比,本发明具有以下技术效果:
[0048]
1、本发明采用三氯异氰尿酸作为合成关键中间体α-氯代乙酰基-γ-丁内酯的氯化剂,氯化反应条件温和,氯化定位选择性好,产物收率和纯度高,不经提纯可直接应用下步中间体合成。简化工艺操作,降低最终产品硫噻唑成本。
[0049]
2、三氯异氰尿酸作为氯化剂,生成的副产物为难溶性白色固体,可直接从反应混合物中过滤分离和回收,作为生产三氯异氰尿酸原料循环回收利用。氯化过程中不产生副产物氯化氢或盐酸,降低三废排放,属于绿色氯化工艺。
[0050]
3、与最常用的氯化剂cl2相比,三氯异氰尿酸稳定性好,刺激性小,毒性低,安全危险性小,操作方便易储存。因此,使用三氯异氰尿酸作为关键中间体α-氯代乙酰基-γ-丁内酯的氯化试剂,该路线适合工业化放大生产。
具体实施方式
[0051]
下面结合具体实施方式对本发明进一步说明。
[0052]
实施例1
[0053]
称取38.4gα-乙酰基-γ-丁内酯,依次加入到安装有搅拌、温度计和冷凝回流的反应瓶中内,搅拌均匀,保持温度在10℃,向其中加入25.2g三氯异氰尿酸,加热反应至30℃持续搅拌2小时,停止反应。冷却至室温,然后过滤反应混合液,滤液为氯代粗产品。减压蒸馏粗产品得到含量为98.1%的α-氯代乙酰基-γ-丁内酯46.3g,收率93.2%。所制备的α-氯代乙酰基-γ-丁内酯的核磁数据如下:1h nmr(600mhz,cdcl3):2.483(m,1h),2.572(s,3h),3.214(m,1h),4.385(m,1h),4.454(m,1h)。
[0054]
称取40.5gα-氯代乙酰基-γ-丁内酯,30ml质量分数5%的稀硫酸,依次加入到安装有搅拌、温度计和冷凝回流反应瓶中内,搅拌下加热反应混合物至100℃,搅拌回流反应3h,停止反应。冷却反应混合液后,用质量分数为5%的naoh水溶液调节反应混合液的ph至7-8。静置反应混合液,分出有机相。水相用适量的二氯甲烷萃取三次,萃取液与有机相混合。混合液经干燥后,脱去溶剂,再减压蒸馏得到含量为97.9%的3-氯-4-氧-1-戊醇产品32.3g,收率92.6%。所制备的3-氯-4-氧-1-戊醇的核磁数据如下:hrms esi(m/z):[m h]
calcd.for c5h
10
clo
2
:137.0364,found:137.0365。
[0055]
称取27.0g 3-氯-4-氧-1-戊醇和30ml 5%稀硫酸,依次加入到安装有搅拌、温度计和冷凝回流器的反应瓶中内,保持低温,搅拌均匀,向其中缓慢滴加20g硫代甲酰胺,加热反应至温度110℃,持续搅拌3小时,至反应完全,冷却反应液至室温。用质量分数为5%的naoh水溶液调节反应混合液的ph至9。将反应混合液静置,分离出有机层。水相用适量二氯甲烷萃取3次,萃取液与有机相混合。干燥混合有机相,脱去溶剂二氯甲烷后,经减压(0.1mpa)蒸馏,收集109-111℃馏分,得到4-甲基-5-(β羟乙基)-噻唑产品,共计26.4g,气相色谱分析含量为92.7%,收率86.5%。hrms esi(m/z):[m h]
calcd.for c6h
10
nos
:144.0478,found:144.0480.
[0056]
实施例2
[0057]
称取39gα-乙酰基-γ-丁内酯溶于100ml二氯甲烷,依次加入到安装有搅拌、温度计、冷凝回流的反应瓶中内,搅拌均匀,保持温度在10℃,向其中28.0g三氯异氰尿酸,加热反应至30℃持续搅拌2小时,至反应完全,过滤,脱除溶剂,气相色谱分析α-氯代乙酰基-γ-丁内酯粗产品含量为88.5%,52.8g,收率94.4%。所制备的α-氯代乙酰基-γ-丁内酯的核磁数据如下:1h nmr(600mhz,cdcl3):2.483(m,1h),2.572(s,3h),3.214(m,1h),4.385(m,1h),4.454(m,1h)。
[0058]
称取上述40.5gα-氯代乙酰基-γ-丁内酯粗产品,45ml质量分数5%的稀硫酸,依次加入到安装有搅拌、温度计和冷凝回流反应瓶中内,搅拌下加热反应至混合物110℃,搅拌回流反应5h,气相色谱检测至α-氯代乙酰基-γ-丁内酯完全反应。冷却反应混合液后,用质量分数为5%的naoh水溶液调节反应混合液的ph至7-8。静置反应混合液,分出有机相。水相用适量的二氯甲烷萃取三次,萃取液与有机相混合。混合液经干燥后,脱去溶剂,再减压蒸馏得到3-氯-4-氧-1-戊醇产品,气相色谱分析3-氯-4-氧-1-戊醇粗产品含量85.2%,32.3g,收率91.4%。
[0059]
称取上述27.0g 3-氯-4-氧-1-戊醇粗产品和50ml 5%稀硫酸,依次加入到安装有
搅拌、温度计和冷凝回流器的反应瓶中内,保持低温,搅拌均匀,向其中缓慢滴加13g硫代甲酰胺,加热反应至温度100℃,持续搅拌3小时,至反应完全,冷却反应液至室温。用质量分数为5%的naoh水溶液调节反应混合液的ph至9。将反应混合液静置,分离出有机层。水相用适量二氯甲烷萃取3次,萃取液与有机相混合。干燥混合有机相,脱去溶剂二氯甲烷后,经减压(0.1mpa)蒸馏,收集109-111℃馏分,得到4-甲基-5-(β羟乙基)-噻唑产品,共计25.4g,气相色谱分析含量为91.5%,收率96.4%。
[0060]
实施例3
[0061]
称取39gα-乙酰基-γ-丁内酯溶于100ml 1,2-二氯甲烷,依次加入到安装有搅拌、温度计、冷凝回流的反应瓶中内,搅拌均匀,保持温度在10℃,向其中28g三氯异氰尿酸,加热反应至30℃持续搅拌2小时,至反应完全,过滤,脱除溶剂,气相色谱分析含量为87.1%,得α-氯代乙酰基-γ-丁内酯粗产品51.9g,收率91.3%。所制备的α-氯代乙酰基-γ-丁内酯的核磁数据如下:1h nmr(600mhz,cdcl3):2.483(m,1h),2.572(s,3h),3.214(m,1h),4.385(m,1h),4.454(m,1h)。
[0062]
称取上述40.5gα-氯代乙酰基-γ-丁内酯粗产品,30ml质量分数10%的稀硫酸,依次加入到安装有搅拌、温度计和冷凝回流反应瓶中内,搅拌下加热反应至混合物100℃,搅拌回流反应4h,气相色谱检测至α-氯代乙酰基-γ-丁内酯完全反应。冷却反应混合液后,用质量分数为5%的naoh水溶液调节反应混合液的ph至7-8。静置反应混合液,分出有机相。水相用适量的二氯甲烷萃取三次,萃取液与有机相混合。混合液经干燥后,脱去溶剂,再减压蒸馏得到3-氯-4-氧-1-戊醇产品,气相色谱分析3-氯-4-氧-1-戊醇粗产品含量72.6%,33.1g,收率81.1%。
[0063]
称取上述27.0g 3-氯-4-氧-1-戊醇粗产品和100ml 5%稀硫酸,依次加入到安装有搅拌、温度计和冷凝回流器的反应瓶中内,保持低温,搅拌均匀,向其中缓慢滴加13g硫代甲酰胺,加热反应至温度100℃,持续搅拌3小时,至反应完全,冷却反应液至室温。用质量分数为5%的naoh水溶液调节反应混合液的ph至9。将反应混合液静置,分离出有机层。水相用适量二氯甲烷萃取3次,萃取液与有机相混合。干燥混合有机相,脱去溶剂二氯甲烷后,经减压(0.1mpa)蒸馏,收集109-111℃馏分,得到4-甲基-5-(β羟乙基)-噻唑产品,共计20.4g,气相色谱分析含量为92.8%,收率92.1%。
[0064]
实施例4
[0065]
称取38.4gα-乙酰基-γ-丁内酯,依次加入到安装有搅拌、温度计和冷凝回流的反应瓶中内,搅拌均匀,保持温度在10℃,向其中加入25.2g三氯异氰尿酸,加热反应至50℃持续搅拌4小时,检测原料α-乙酰基-γ-丁内酯至反应完全。过滤反应混合液,滤液为氯代粗产品。减压蒸馏粗产品得到含量为97.8%的α-氯代乙酰基-γ-丁内酯33.4g,收率67.0%。所制备的α-氯代乙酰基-γ-丁内酯的核磁数据如下:1h nmr(600mhz,cdcl3):2.483(m,1h),2.572(s,3h),3.214(m,1h),4.385(m,1h),4.454(m,1h)。
[0066]
取32.5g前步合成的α-氯代乙酰基-γ-丁内酯,50ml质量分数5%的稀硫酸,依次加入到安装有搅拌、温度计和冷凝回流反应瓶中内,搅拌下加热反应混合物至100℃,搅拌回流反应8h,气相色谱检测至α-氯代乙酰基-γ-丁内酯完全反应。冷却反应混合液后,用质量分数为5%的naoh水溶液调节反应混合液的ph至7-8。静置反应混合液,分出有机相。水相用适量的二氯甲烷萃取三次,萃取液与有机相混合。混合液经干燥后,脱去溶剂,再减压蒸
馏得到含量为92.1%的3-氯-4-氧-1-戊醇产品20.6g,收率71.0%。
[0067]
称取17.6g前一步合成的3-氯-4-氧-1-戊醇和50ml 5%稀硫酸,依次加入到安装有搅拌、温度计和冷凝回流器的反应瓶中内,保持低温,搅拌均匀,向其中缓慢滴加14g硫代甲酰胺,加热反应至温度110℃,持续搅拌3小时,至反应完全,冷却反应液至室温。用质量分数为5%的naoh水溶液调节反应混合液的ph至9。将反应混合液静置,分离出有机层。水相用适量二氯甲烷萃取3次,萃取液与有机相混合。干燥混合有机相,脱去溶剂二氯甲烷后,经减压(0.1mpa)蒸馏,收集109-111℃馏分,得到4-甲基-5-(β羟乙基)-噻唑产品,共计15.8g,气相色谱分析含量为92.3%,收率86.6%。
[0068]
实施例5
[0069]
称取25.2g三氯异氰尿酸和39gα-乙酰基-γ-丁内酯,依次加入到安装有搅拌、温度计、冷凝回流的反应瓶中内,搅拌均匀,加热反应至30℃持续搅拌2小时,过滤。而后向滤液中加入35ml质量分数5%的稀硫酸,加热反应至温度为110℃,持续搅拌3小时后,取样检测反应进度至完全,补加100ml 5%稀硫酸和17g硫代甲酰胺,持续搅拌4小时,至反应完全,冷却反应液至室温,用质量分数为5%的naoh水溶液条件ph至9,过滤,分离出有机层,并用二氯甲烷萃取有机层3次,萃取液经减压(0.1mpa)蒸馏,收集109-111℃,得4-甲基-5-(β羟乙基)-噻唑产品28.7g,色谱分析含量为93.4%,收率61.5%。
[0070]
以上所述之实施例,只是本发明的较佳实施例而已,仅仅用以解释本发明,并非限制本发明实施范围,对于本技术领域的技术人员来说,当然可根据本说明书中所公开的技术内容,通过置换或改变的方式轻易做出其它的实施方式,故凡在本发明的原理及工艺条件所做的变化和改进等,均应包括于本发明申请专利范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。