用于抗肿瘤疗法中的双重atm和dna-pk抑制剂
技术领域
1.本发明涉及作为单一疗法或与放射疗法、化学疗法和/或免疫疗法组合用于治疗癌症的化合物及其药学上可接受的盐以及它们的使用方法。
2.发明背景
3.丝氨酸-苏氨酸激酶的pikk(pi-3k样激酶)家族的几个成员是已知的dna损伤信号传导的介体。
4.放射疗法(rt)用于在其患病期间的某个时间点治疗》50%的所有癌症患者。尽管作出了很大的努力,但以前开发临床放射增敏剂的方法并不是非常有效,主要是由于靶向非特异性途径的结果,该途径不是对放射的细胞响应的直接调节因子。
5.需要针对肿瘤疾病的新疗法。
技术实现要素:
6.一般来说,本发明提供一种式(i)的化合物:
[0007][0008]
或其药学上可接受的盐,
[0009]
其中
[0010]
z是ch、cr3或n;
[0011]
y是chr5或nr6;
[0012]
n是0、1、2或3;
[0013]
r1是
–o–
l
–
n(r7)2或任选取代的四元饱和n-杂环基;
[0014]
r2是c
1-3
烷基;
[0015]
每个r3独立地是卤素;
[0016]
r4是任选取代的烷基;
[0017]
r5是氢、任选取代的c
1-3
烷基或苄氧基;
[0018]
r6是任选取代的c
1-3
烷基;
[0019]
每个r7独立地是h或任选取代的c
1-3
烷基;且
[0020]
l是任选取代的亚乙基。
[0021]
在一些实施方案中,n是1。在某些实施方案中,所述化合物是式(ia)的化合物:
[0022][0023]
或其药学上可接受的盐。
[0024]
在具体实施方案中,r3是卤素(例如氟)。在其他实施方案中,n是0。
[0025]
在又其他实施方案中,所述化合物是式(ib)的化合物:
[0026][0027]
或其药学上可接受的盐。
[0028]
在仍其他实施方案中,r1是
–o–
l
–
n(r7)2。在一些实施方案中,一个r7是h,且其余r7任选地是c
1-3
烷基。在某些实施方案中,至少一个r7是异丙基。在具体实施方案中,r2是甲基、乙基或异丙基。在其他实施方案中,r2是甲基。在又其他实施方案中,r4是甲基。在又其他实施方案中,y是chr5。在仍其他实施方案中,r5是氢。在其他实施方案中,r5是任选取代的c
1-3
烷基。在又其他实施方案中,r5是苄氧基。在仍其他实施方案中,y是nr6。在一些实施方案中,r6是任选取代的c3烷基。在某些实施方案中,r6是异丙基。
[0029]
在具体实施方案中,所述化合物选自由以下组成的组:
[0030]
[0031][0032]
及其药学上可接受的盐。
[0033]
在其他实施方案中,所述化合物具有以下结构:
[0034]
或其药学上可接受的盐。
[0035]
在又其他实施方案中,所述化合物具有以下结构:
[0036]
或其药学上可接受的盐。
[0037]
在仍其他实施方案中,所述化合物具有以下结构:
[0038][0039]
或其药学上可接受的盐。
[0040]
另一方面,本发明提供一种药物组合物,其包含本发明化合物或其药学上可接受的盐和药学上可接受的赋形剂。
[0041]
又一方面,本发明提供一种治疗肿瘤疾病(例如,癌症,例如本文所述的那些癌症)
的方法,其通过向有此需要的患者施用治疗有效量的本发明化合物或其药学上可接受的盐或本发明的药物组合物来进行。仍另一方面,本发明提供药物组合物,其是用于治疗肿瘤疾病(例如,癌症,例如本文所述的那些癌症)。所述药物组合物包含本发明化合物。再一方面,本发明提供本发明化合物在制造供治疗肿瘤疾病(例如,癌症,例如本文所述的那些癌症)用的药剂中的用途。在一些实施方案中,所述肿瘤疾病是脑癌、膀胱癌、乳腺癌、中枢神经系统癌、宫颈癌、结肠癌、子宫内膜癌、食道癌、胃肠道间质瘤、胃癌、头颈癌、颊癌、口腔癌、肝细胞癌、肺癌、黑色素瘤、梅克尔(merkel cell carcinoma)细胞癌、间皮瘤、鼻咽癌、神经母细胞瘤、骨肉瘤、卵巢癌、胰腺癌、前列腺癌、肾癌、唾液腺癌、肉瘤、睾丸癌、尿路上皮癌、外阴癌或威尔姆氏瘤(wilm’s tumor)。在其他实施方案中,所述肿瘤疾病是乳腺癌、肺癌、头颈癌、胰腺癌、直肠癌、胶质母细胞瘤、肝细胞癌、胆管癌、转移性肝脏病变、黑色素瘤、骨肉瘤、软组织肉瘤、子宫内膜癌、宫颈癌、前列腺癌或梅克尔细胞癌。
[0042]
在一些实施方案中,患者正在接受放射疗法。在某些实施方案中,所述化合物或所述药物组合物与放射疗法同时施用于患者。在具体实施方案中,所述化合物或所述药物组合物在放射疗法之前施用于患者。在其他实施方案中,所述化合物或所述药物组合物在放射疗法之后施用于患者。在又其他实施方案中,放射疗法包括外部、内部、近距放射疗法或全身性暴露,例如用放射性核素(例如,β-发射放射性核素(例如,
32
磷、
67
铜、
77
溴、
89
锶、
90
钇、
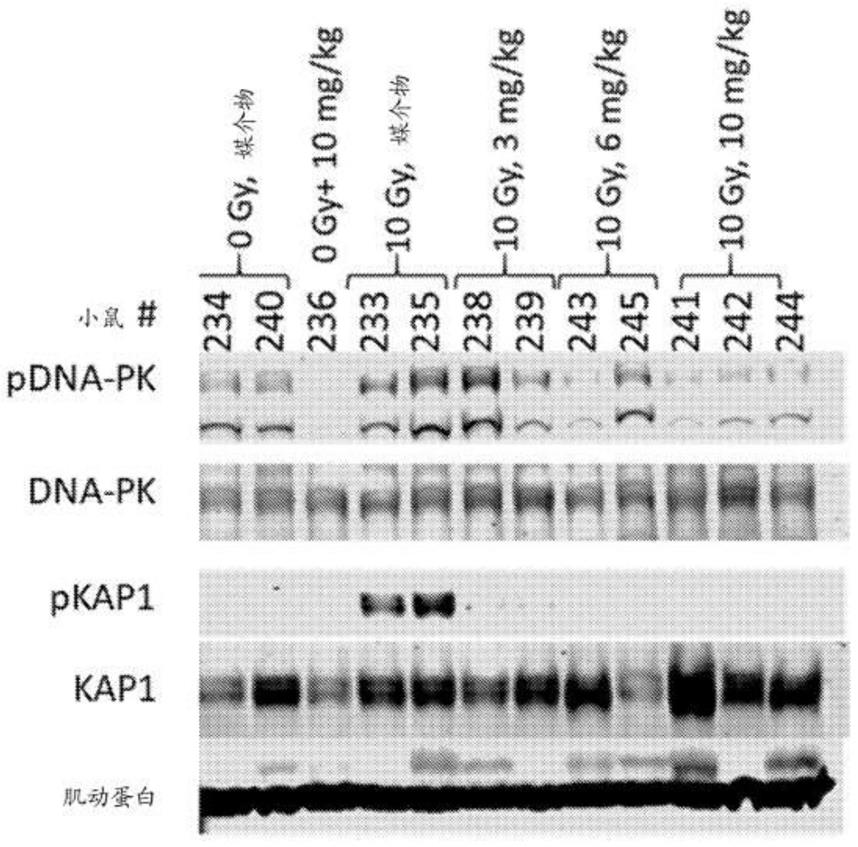
105
铑、
131
碘、
137
铯、
149
钷、
153
钐、
166
钬、
177
镥、
186
铼、
188
铼或
199
金)、α-发射放射性核素(例如,
211
砹、
213
铋、
223
镭、
225
锕或
227
钍)、γ射线发射放射性核素(例如,
192
铱)或电子俘获放射性核素(例如,
67
镓、
103
钯或
125
碘))、抗体放射性核素缀合物(例如,
90
y-替伊莫单抗(ibritumomab tiuxetane)、
131
i-托西莫单抗、
225
ac-沙林妥珠单抗(lintuzumab satetraxetan)、
227
th-柯安妥单抗(anetumab corixetan)、
90
y-西依匹莫单抗(epitumomab cituxeta n)、
90
y-替坦-克利妥珠单抗(clivatuzumab tetraxetan)、
177
lu-沙泰坦-利洛托单抗(lilotomab satetraxetan)、
90
y-替坦-罗索帕他单抗(rosopata mab tetraxetan)、
90
y-巴坦比妥昔单抗(tabituximab barzuxetan)或
90
y-替坦-他珠单抗(tacatuzumab tetraxetan))或另一种靶向放射性核素缀合物(例如,
131
i-psma、
90
y-psma、
177
lu-psma或
177
lu-替坦-沙托肽(satoreotide tetraxetan))。优选地,放射疗法包括施用抗体放射性核素缀合物。在仍其他实施方案中,患者正在接受抗肿瘤剂。在其他实施方案中,所述抗肿瘤剂是顺铂、奥沙利铂、卡铂、戊柔比星、伊达比星、卡里奇霉素或parp抑制剂中的一种或多种。在又其他实施方案中,所述抗肿瘤剂是抗肿瘤生物剂和/或抗肿瘤免疫治疗剂。在仍其他实施方案中,所述化合物或所述药物组合物与所述抗肿瘤剂同时施用于患者。在一些实施方案中,所述化合物或所述药物组合物在所述抗肿瘤剂之前施用于患者。在某些实施方案中,所述化合物或所述药物组合物在所述抗肿瘤剂之后施用于患者。
具体实施方式
[0043]
定义
[0044]
应当理解的是,本文采用的术语用于描述具体实施方案的目的,而不是意在限制。进一步,尽管与本文所述的那些相似或等效的任何方法、装置和材料可用于本发明的实践或测试中,但现在描述优选的方法、装置和材料。除了上述内容以外,如说明书和所附权利要求中所使用,除非相反地指出,否则以下术语具有所示的含义:
[0045]“氨基”是指-nh2基团。
[0046]“氰基”是指-cn基团。
[0047]“羟基”是指-oh基团。
[0048]“亚氨基”是指=nh取代基。
[0049]“硝基”是指-no2基团。
[0050]“氧代”是指=o取代基。
[0051]“硫代”是指=s取代基。
[0052]“三氟甲基”是指-cf3基团。
[0053]“烷基”是指具有1至12个碳原子、优选1至8个碳原子或1至6个碳原子且通过单键连接至分子的剩余部分的直链、饱和、无环、单价烃基或支链、饱和、无环、单价烃基,例如甲基、乙基、正丙基、1-甲基乙基(异丙基)、正丁基、正戊基、1,1-二甲基乙基(叔丁基)、3-甲基己基、2-甲基己基等。任选取代的烷基是任选地被1、2、3、4或5个取代基取代(价态允许)的烷基,所述取代基独立地选自由以下组成的组:卤基、氰基、硝基、芳基、环烷基、杂环基、杂芳基、氧代、三甲基硅烷基、-or
14
、-oc(o)-r
14
、-n(r
14
)2、-c(o)r
15
、-c(o)or
14
、-c(o)n(r
14
)2、-n(r
14
)c(o)or
16
、-n(r
14
)c(o)r
16
、-n(r
14
)s(o)
tr16
(其中t是1或2)、-s(o)
t
or
16
(其中t是1或2)、-s(o)
pr16
(其中p是0、1或2)和-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、环烷基烷基、芳基、芳烷基、杂环基或杂芳基;每个r
15
独立地是氢、环烷基、芳基、杂环基或杂芳基;并且每个r
16
独立地是烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基。
[0054]“烯基”是指含有1、2或3个碳-碳双键、具有2至12个碳原子、优选2至8个碳原子且通过单键连接至分子的剩余部分的直链、无环、单价烃基或支链、无环、单价烃基,例如乙烯基、丙-1-烯基、丁-1-烯基、戊-1-烯基、戊-1,4-二烯基等。任选取代的烯基是任选地被1、2、3、4或5个取代基取代(价态允许)的烯基,所述取代基独立地选自由以下组成的组:卤基、氰基、硝基、芳基、环烷基、杂环基、杂芳基、氧代、三甲基硅烷基、-or
14
、-oc(o)-r
14
、-n(r
14
)2、-c(o)r
15
、-c(o)or
14
、-c(o)n(r
14
)2、-n(r
14
)c(o)or
16
、-n(r
14
)c(o)r
16
、-n(r
14
)s(o)
tr16
(其中t是1或2)、-s(o)
t
or
16
(其中t是1或2)、-s(o)
pr16
(其中p是0、1或2)和-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是氢、环烷基、芳基、杂环基或杂芳基;并且每个r
16
独立地是烷基、卤代烷基、环烷基、环烷基烷基、芳基、杂环基或杂芳基。
[0055]“炔基”是指含有1或2个碳-碳三键和任选地1、2或3个碳-碳双键且具有2至12个碳原子、优选2至8个碳原子且通过单键连接至分子的剩余部分的直链、无环、单价烃基或支链、无环、单价烃基,例如乙炔基、丙-1-炔基、丁-1-炔基、戊-1-炔基、戊-1-烯4-炔基等。任选取代的炔基是任选地被1、2、3、4或5个取代基取代的炔基,所述取代基独立地选自由以下组成的组:卤基、氰基、硝基、芳基、环烷基、杂环基、杂芳基、氧代、三甲基硅烷基、-or
14
、-oc(o)-r
14
、-n(r
14
)2、-c(o)r
15
、-c(o)or
14
、-c(o)n(r
14
)2、-n(r
14
)c(o)or
16
、-n(r
14
)c(o)r
16
、-n(r
14
)s(o)
tr16
(其中t是1或2)、-s(o)
t
or
16
(其中t是1或2)、-s(o)
pr16
(其中p是0、1或2)和-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是氢、环烷基、芳基、杂环基或杂芳基;并且每个r
16
独立地是烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基。
[0056]“亚烷基”或“亚烷基链”是指具有1至12个碳原子的直链、无环、饱和、二价烃链或支链、无环、饱和、二价烃链,例如亚甲基、亚乙基、亚丙基、正亚丁基等。亚烷基链通过单键连接。亚烷基链的连接点可以在亚烷基链内的同一碳原子上或不同碳原子上。任选取代的亚烷基链是任选地被1、2、3、4或5个取代基取代(价态允许)的亚烷基链,所述取代基独立地选自由以下组成的组:卤基、氰基、硝基、芳基、环烷基、杂环基、杂芳基、氧代、三甲基硅烷基、-or
14
、-oc(o)-r
14
、-n(r
14
)2、-c(o)r
15
、-c(o)or
14
、-c(o)n(r
14
)2、-n(r
14
)c(o)or
16
、-n(r
14
)c(o)r
16
、-n(r
14
)s(o)
tr16
(其中t是1或2)、-s(o)
t
or
16
(其中t是1或2)、-s(o)
pr16
(其中p是0、1或2)和-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是氢、环烷基、芳基、杂环基或杂芳基;并且每个r
16
独立地是烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基。在一些实施方案中,亚烷基是亚乙基。
[0057]“亚烯基”或“亚烯基链”是指含有1、2或3个碳-碳双键且具有2至12个碳原子的直链、无环、二价烃链或支链、无环、二价烃链,例如,亚乙烯基、亚丙烯基、正亚丁烯基等。亚烯基链通过单键连接。亚烯基链的连接点可以在亚烯基链内的同一碳原子上或不同碳原子上。任选取代的亚烯基链是任选地被1、2、3、4或5个取代基取代(价态允许)的亚烯基链,所述取代基独立地选自由以下组成的组:卤基、氰基、硝基、芳基、环烷基、杂环基、杂芳基、氧代、三甲基硅烷基、-or
14
、-oc(o)-r
14
、-n(r
14
)2、-c(o)r
15
、-c(o)or
14
、-c(o)n(r
14
)2、-n(r
14
)c(o)or
16
、-n(r
14
)c(o)r
16
、-n(r
14
)s(o)
tr16
(其中t是1或2)、-s(o)
t
or
16
(其中t是1或2)、-s(o)
pr16
(其中p是0、1或2)和-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是氢、环烷基、芳基、杂环基或杂芳基;并且每个r
16
独立地是烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基。
[0058]“亚炔基”或“亚炔基链”是指含有1或2个碳-碳三键和任选地1、2或3个碳-碳双键且具有2至12个碳原子的直链、无环、二价烃链或支链、无环、二价烃链,例如亚丙炔基、正亚丁炔基等。亚炔基链通过单键连接。亚炔基的连接点可以在亚炔基链内的同一碳原子上或不同碳原子上。任选取代的亚炔基链是任选地被1、2、3、4或5个取代基取代的亚炔基链,所述取代基独立地选自由以下组成的组:卤基、氰基、硝基、芳基、环烷基、杂环基、杂芳基、氧代、三甲基硅烷基、-or
14
、-oc(o)-r
14
、-n(r
14
)2、-c(o)r
15
、-c(o)or
14
、-c(o)n(r
14
)2、-n(r
14
)c(o)or
16
、-n(r
14
)c(o)r
16
、-n(r
14
)s(o)
tr16
(其中t是1或2)、-s(o)
t
or
16
(其中t是1或2)、-s(o)
pr16
(其中p是0、1或2)和-s(o)
t
n(r
14
)2(其中t是1至2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是氢、环烷基、芳基、杂环基或杂芳基;并且每个r
16
独立地是烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基。
[0059]“烷氧基”是指式-ora的基团,其中ra是含有1至12个碳原子的如上述定义的烷基。任选取代的烷氧基的烷基部分任选地如上文关于烷基所定义的那样被取代。
[0060]“烷氧基烷基”是指式-r
a-o-rb的基团,其中ra是亚烷基且rb是如上所定义的烷基。任选取代的烷氧基烷基的烷基和亚烷基部分分别任选地如上文关于烷基和亚烷基链所定义的那样被取代。
[0061]“芳烷基”是指式-r
a-rb的基团,其中ra是亚烷基且rb是如本文所述的芳基。任选取代的芳烷基的亚烷基和芳基部分分别任选地如本文关于亚烷基和芳基所述的那样被取代。
[0062]“芳基”是指含有6至18个碳原子的芳族单环或多环烃环系统基团,其中多环芳环系统是双环、三环或四环环系统。芳基包括但不限于诸如芴基、苯基和萘基的基团。任选取
代的芳基是任选地被1、2、3、4或5个取代基取代的芳基,所述取代基独立地选自由以下组成的组:烷基、烯基、卤基、卤代烷基、卤代烯基、氰基、硝基、芳基、杂芳基、杂芳基烷基、-r
15-or
14
、-r
15-oc(o)-r
14
、-r
15-n(r
14
)2、-r
15-c(o)r
14
、-r
15-c(o)or
14
、-r
15-c(o)n(r
14
)2、-r
15-n(r
14
)c(o)or
16
、-r
15-n(r
14
)c(o)r
16
、-r
15-n(r
14
)s(o)
tr16
(其中t是1或2)、-r
15-s(o)
t
or
16
(其中t是1或2)、-r
15-s(o)
pr16
(其中p是0、1或2)和-r
15-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是直接键或直链或支链亚烷基或亚烯基链;并且每个r
16
独立地是烷基、卤代烷基、环烷基、环烷基烷基、芳基、杂环基或杂芳基。
[0063]“芳基烷氧基”是指式
–
o-r的基团,其中r是芳烷基。任选被取代的芳基烷氧基是如本文针对芳烷基所述任选被取代的芳基烷氧基。在一些实施方案中,芳基烷氧基是苄氧基。
[0064]“环烷基”是指具有3至15个碳原子、优选具有3至10个碳原子并且是饱和或不饱和的且通过单键连接至分子的剩余部分的稳定的非芳族单环或多环烃基。多环烃基是双环、三环或四环环系统。不饱和环烷基含有1、2或3个碳-碳双键和/或1个碳-碳三键。单环环烷基包括例如环丙基、环丁基、环戊基、环己基、环庚基和环辛基。多环环烷基包括例如金刚烷基、降冰片基、十氢萘基等。任选取代的环烷基是任选地被1、2、3、4或5个取代基取代的环烷基,所述取代基独立地选自由以下组成的组:烷基、烯基、卤基、卤代烷基、卤代烯基、氰基、硝基、氧代、芳基、芳烷基、环烷基、杂环基、杂芳基、-r
15-or
14
、-r
15-oc(o)-r
14
、-r
15-n(r
14
)2、-r
15-c(o)r
14
、-r
15-c(o)or
14
、-r
15-c(o)n(r
14
)2、-r
15-n(r
14
)c(o)or
16
、-r
15-n(r
14
)c(o)r
16
、-r
15-n(r
14
)s(o)
tr16
(其中t是1或2)、-r
15-s(o)
t
or
16
(其中t是1或2)、-r
15-s(o)
pr16
(其中p是0、1或2)和-r
15-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是直接键或直链或支链亚烷基或亚烯基链;并且每个r
16
独立地是烷基、卤代烷基、环烷基、环烷基烷基、芳基、芳烷基、杂环基或杂芳基。
[0065]“稠合的”是指与本发明化合物中的现有环结构稠合的本文所述的任何环系统。当稠合的环系统为杂环基或杂芳基时,变为稠合的环系统的一部分的现有环结构上的任何碳原子可被氮原子替代。
[0066]“卤基”是指卤素取代基:溴、氯、氟和碘。
[0067]“卤代烷基”是指如上所定义的烷基,其进一步被一个或多个卤素取代基取代。卤代烷基中包含的卤基取代基的数目是1至最多达可供用卤基取代基替代的氢原子的总数(例如全氟烷基)。卤代烷基的非限制性实例包括三氟甲基、二氟甲基、三氯甲基、2,2,2-三氟乙基、1-氟甲基-2-氟乙基、3-溴-2-氟丙基、1-溴甲基-2-溴乙基等。对于任选取代的卤代烷基,与卤代烷基的烷基部分的碳原子键合的氢原子可以任选地用如上针对任选取代的烷基所定义的取代基替代。
[0068]“卤代烯基”是指如上所定义的烯基,其进一步被一个或多个卤基取代基取代。卤代烯基中包含的卤基取代基的数目是1至最多达可供用卤基取代基替代的氢原子的总数(例如全氟烯基)。卤代烯基的非限制性实例包括2,2-二氟乙烯基、3-氯丙-1-烯基等。对于任选取代的卤代烯基,与卤代烯基的烯基部分的碳原子键合的氢原子可以任选地用如上针对任选取代的烯基所定义的取代基替代。
[0069]“卤代炔基”是指如上所定义的炔基,其进一步被一个或多个卤基取代基取代。卤代炔基中包含的卤基取代基的数目是1至最多达可供用卤基取代基替代的氢原子的总数
(例如全氟炔基)。卤代炔基的非限制性实例包括3-氯丙-1-炔基等。卤代炔基的炔基部分可以另外任选地如上文关于炔基所定义的那样被取代。
[0070]“杂芳基烷基”是指式-r
a-rb的基团,其中ra是亚烷基且rb是如本文所述的杂芳基。任选取代的杂芳基烷基的亚烷基和杂芳基部分分别任选地如本文关于亚烷基和杂芳基所述的那样被取代。
[0071]“杂环基”是指具有2至12的碳数且总共含有1至6个独立地选自由氮、氧、磷和硫组成的组的杂原子的稳定的3至18元非芳族环系统基团。杂环基是单环、双环、三环或四环环系统。双环、三环或四环杂环基是稠环、螺环和/或桥环系统。杂环基可以是饱和的或不饱和的。不饱和杂环基含有1、2或3个碳-碳双键和/或1个碳-碳三键。任选取代的杂环基是任选地被1、2、3、4或5个取代基取代的杂环基,所述取代基独立地选自由以下组成的组:烷基、烯基、卤基、卤代烷基、卤代烯基、氰基、氧代、硫代、硝基、芳基、芳烷基、环烷基、杂环基、杂芳基、-r
15-or
14
、-r
15-oc(o)-r
14
、-r
15-n(r
14
)2、-r
15-c(o)r
14
、-r
15-c(o)or
14
、-r
15-c(o)n(r
14
)2、-r
15-n(r
14
)c(o)or
16
、-r
15-n(r
14
)c(o)r
16
、-r
15-n(r
14
)s(o)
tr16
(其中t是1或2)、-r
15-s(o)
t
or
16
(其中t是1或2)、-r
15-s(o)
pr16
(其中p是0、1或2)和-r
15-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、烯基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是直接键或直链或支链亚烷基或亚烯基链;并且每个r
16
独立地是烷基、烯基、卤代烷基、环烷基、芳基、杂环基或杂芳基。杂环基中的氮、碳或硫原子可以任选地被氧化(当取代基是氧代且存在于杂原子上时);氮原子可以任选地被季铵化(当取代基是烷基、烯基、芳基、芳烷基、环烷基、杂环基、杂芳基、-r
15-or
14
、-r
15-oc(o)-r
14
、-r
15-n(r
14
)2、-r
15-c(o)r
14
、-r
15-c(o)or
14
、-r
15-c(o)n(r
14
)2、-r
15-n(r
14
)c(o)or
16
、-r
15-n(r
14
)c(o)r
16
、-r
15-n(r
14
)s(o)
tr16
(其中t是1或2)、-r
15-s(o)
t
or
16
(其中t是1或2)、-r
15-s(o)
pr16
(其中p是0、1或2)和-r
15-s(o)
t
n(r
14
)2(其中t是1或2)时,其中r
15
是直链或支链亚烷基或亚烯基链,并且r
14
和r
16
如上所定义)。任选取代的杂环基的实例包括但不限于氮杂环丁烷基、二氧杂环戊烷基、噻吩基[1,3]二噻烷基、十氢异喹啉基、咪唑啉基、咪唑烷基、异噻唑烷基、异噁唑烷基、吗啉基、八氢吲哚基、八氢异吲哚基、2-氧代哌嗪基、2-氧代哌啶基、2-氧代吡咯烷基、噁唑烷基、哌啶基、哌嗪基、4-哌啶酮基、吡咯烷基、吡唑烷基、噻唑烷基、四氢呋喃基、三噻烷基、四氢吡喃基、硫代吗啉基、硫代吗啉基、1-氧代-硫代吗啉基和1,1-二氧代-硫代吗啉基。
[0072]“亚杂环基”是指其中一个氢原子被化合价替代的杂环基。任选取代的亚杂环基任选地如本文针对杂环基所述的那样被取代。
[0073]“杂芳基”是指含有至少一个芳环、具有1至17个碳原子的碳数且含有总共1至10个独立地选自由氮、氧和硫组成的组的杂原子的5至18元环系统基团。杂芳基是单环、双环、三环或四环环系统。双环、三环或四环杂芳基是稠环和/或桥环系统。任选取代的杂芳基是任选地被1、2、3、4或5个取代基取代的杂芳基,所述取代基独立地选自由以下组成的组:烷基、烯基、烷氧基、卤基、卤代烷基、卤代烯基、氰基、氧代、硫代、硝基、氧代、芳基、芳烷基、环烷基、杂环基、杂芳基或杂芳基烷基、-r
15-or
14
、-r
15-oc(o)-r
14
、-r
15-n(r
14
)2、-r
15-c(o)r
14
、-r
15-c(o)or
14
、-r
15-c(o)n(r
14
)2、-r
15-n(r
14
)c(o)or
16
、-r
15-n(r
14
)c(o)r
16
、-r
15-n(r
14
)s(o)
tr16
(其中t是1或2)、-r
15-s(o)
t
or
16
(其中t是1或2)、-r
15-s(o)
tr16
(其中p是0、1或2)和-r
15-s(o)
t
n(r
14
)2(其中t是1或2),其中每个r
14
独立地是氢、烷基、烯基、卤代烷基、环烷基、芳基、杂环基或杂芳基;每个r
15
独立地是直接键或直链或支链亚烷基或亚烯基链;并且每个r
16
是
烷基、烯基、卤代烷基、环烷基、芳基、杂环基或杂芳基。杂环基中的氮、碳或硫原子可以任选地被氧化(当取代基是氧代且存在于杂原子上时),条件是杂芳基中的至少一个环保持芳族;氮原子可以任选地被季铵化(当取代基是烷基、烯基、芳基、芳烷基、环烷基、杂环基、杂芳基、-r
15-or
14
、-r
15-oc(o)-r
14
、-r
15-n(r
14
)2、-r
15-c(o)r
14
、-r
15-c(o)or
14
、-r
15-c(o)n(r
14
)2、-r
15-n(r
14
)c(o)or
16
、-r
15-n(r
14
)c(o)r
16
、-r
15-n(r
14
)s(o)
tr16
(其中t是1或2)、-r
15-s(o)
t
or
16
(其中t是1或2)、-r
15-s(o)
pr16
(其中p是0、1或2)和-r
15-s(o)
t
n(r
14
)2(其中t是1或2)时,其中r
15
是直链或支链亚烷基或亚烯基链,并且r
14
和r
16
如上所定义),条件是杂芳基中的至少一个环保持芳族。任选取代的杂芳基基团的实例包括、但不限于氮杂基、吖啶基、苯并咪唑基、苯并噻唑基、苯并吲哚基、苯并间二氧杂环戊烯基、苯并呋喃基、苯并噁唑基、苯并噻唑基、苯并噻二唑基、苯并[b][1,4]二氧杂基、1,4-苯并二噁烷基、苯并萘并呋喃基、苯并噁唑基、苯并间二氧杂环戊烯基、苯并二氧杂环己烯基(benzodioxinyl)、苯并吡喃基、苯并吡喃酮基、苯并呋喃基、苯并呋喃酮基、苯并噻吩基(苯并噻吩基)、苯并三唑基、苯并[4,6]咪唑并[1,2-a]吡啶基、咔唑基、噌啉基、二苯并呋喃基、二苯并噻吩基、呋喃基、呋喃酮基、异噻唑基、咪唑基、吲唑基、吲哚基、吲唑基、异吲哚基、吲哚啉基、异吲哚啉基、异喹啉基、吲嗪基、异噁唑基、萘基、萘啶基、噁二唑基、2-氧代氮杂基、噁唑基、环氧乙烷基、1-苯基-1h-吡咯基、吩嗪基、吩噻嗪基、吩噁嗪基、酞嗪基、蝶啶基、嘌呤基、吡咯基、吡唑基、吡啶基、吡嗪基、嘧啶基、哒嗪基、吡咯基、喹唑啉基、喹喔啉基、喹啉基、奎宁环基、异喹啉基、四氢喹啉基、噻唑基、噻二唑基、三唑基、四唑基、三嗪基和硫代苯基基(即,噻吩基)。
[0074]
本文公开的发明还意在涵盖式(i)的所有药学上可接受的化合物,其通过使一个或多个原子被具有不同原子质量或质量数的原子替代而被同位素标记。可掺入所公开化合物中的同位素的实例分别包括氢、碳、氮、氧、磷、氟、氯和碘的同位素,诸如2h、3h、
11
c、
13
c、
14
c、
13
n、
15
n、
15
o、
17
o、
18
o、
31
p、
32
p、
35
s、
18
f、
36
cl、
123
i和
125
i。这些放射性标记的化合物可用于通过表征例如atm和dna-pk酶上的作用位点或模式,或与atm和dna-pk酶上的药理学上重要的作用位点的结合亲和力而帮助测定或测量化合物的有效性。式(i)的某些同位素标记的化合物,例如,掺入放射性同位素的那些,适用于药物和/或底物组织分布研究。鉴于它们易于掺入和易于检测的手段,放射性同位素氚(即,3h)和碳14(即,
14
c)特别适用于此目的。
[0075]
用较重同位素(诸如氘,即,2h)取代可提供由较大的代谢稳定性(例如,提高的体内半衰期)或降低的剂量需求得到的某些治疗优势,且因此,在一些情况下可以是优选的。
[0076]
用正电子发射同位素(诸如
11
c、
18
f、
15
o和
13
n)取代可用于正电子发射断层显像(pet)研究,用于检查底物受体占用率。使用适当的同位素标记的试剂替代先前采用的非标记的试剂,式(i)的同位素标记的化合物通常可通过本领域技术人员已知的常规技术或通过与在下文所列出的实施例和制备中描述的那些类似的方法来制备。
[0077]
本文公开的发明还意在涵盖所公开化合物的体内代谢产物。此类产物可例如由施用的化合物的氧化、还原、水解、酰胺化、酯化等而产生,主要是由于酶促过程。因此,本发明包括通过一种方法产生的化合物,所述方法包括使本发明化合物与哺乳动物接触足以得到其代谢产物的一段时间。此类产物通常通过以可检测的剂量将本发明的放射性标记的化合物施用于动物(诸如大鼠、小鼠、豚鼠、犬、猴)或人,允许足够的时间使得发生代谢,并且将其转化产物从尿、血液或其它生物学样品分离而鉴定。
[0078]“稳定的化合物”和“稳定的结构”意在指示足够坚固以经受从反应混合物分离至
可用的纯度并配制成有效治疗剂的化合物。
[0079]“哺乳动物”包括人,以及家养动物(诸如实验室动物和家庭宠物(例如猫、狗、猪、牛、绵羊、山羊、马、兔)和非家养动物(诸如野生动物)等。
[0080]“任选的”或“任选(地)”意味着随后描述的情况的事件可能发生或者可能不发生,并且该描述包括其中所述事件或情况发生的例子以及其中其不发生的例子。例如,“任选取代的芳基”意味着芳基可被取代或可不被取代,并且该描述包括取代的芳基和不具有取代的芳基两者。
[0081]“患者”意指患有疾病或病状的人或非人动物(例如哺乳动物),由合格的专业人员(例如,医生、执业护士或兽医)在有或没有本领域中已知的来自患者的样品的实验室测试下确定。
[0082]“药学上可接受的载体、稀释剂或赋形剂”包括但不限于已通过美国食品和药品管理局批准可被接受用于人或家养动物的任何助剂、载体、赋形剂、助流剂、增甜剂、稀释剂、防腐剂、染料/着色剂、香味增强剂、表面活性剂、湿润剂、分散剂、悬浮剂、稳定剂、等渗剂、溶剂或乳化剂。
[0083]
如本文所用,“药学上可接受的盐”表示在合理的医学判断范围内,适用于与人和动物的组织接触而没有不当毒性、刺激、过敏反应等并且与合理的益处/风险比相称的那些盐。药学上可接受的盐是本领域中公知的。例如,药学上可接受的盐描述于以下文献中:berge等人,j.pharmaceutical sciences 66:1-19,1977及pharmaceutical salts:properties,selection,and use,(编者p.h.stahl和c.g.wermuth),wiley-vch,2008。药学上可接受的盐包括酸和碱加成盐。
[0084]“药学上可接受的酸加成盐”是指保持游离碱的生物学有效性和特性的那些盐,其不会在生物学上或其它方面不合需要,并且其用无机酸(诸如但不限于盐酸、氢溴酸、硫酸、硝酸、磷酸等)和有机酸(诸如但不限于乙酸、2,2-二氯乙酸、己二酸、海藻酸、抗坏血酸、天冬氨酸、苯磺酸、苯甲酸、4-乙酰氨基苯甲酸、樟脑酸、樟脑-10-磺酸、癸酸、己酸、辛酸、碳酸、肉桂酸、柠檬酸、环拉酸、十二烷基硫酸、乙烷-1,2-二磺酸、乙磺酸、2-羟基乙磺酸、甲酸、富马酸、半乳糖二酸、龙胆酸、葡庚糖酸、葡糖酸、葡糖醛酸、谷氨酸、戊二酸、2-氧代-戊二酸、甘油磷酸、羟基乙酸、马尿酸、异丁酸、乳酸、乳糖酸、月桂酸、马来酸、苹果酸、丙二酸、扁桃酸、甲磺酸、粘液酸、萘-1,5-二磺酸、萘-2-磺酸、1-羟基-2-萘甲酸、烟酸、油酸、乳清酸、草酸、棕榈酸、扑酸、丙酸、焦谷氨酸、丙酮酸、水杨酸、4-氨基水杨酸、癸二酸、硬脂酸、琥珀酸、酒石酸、硫氰酸、对甲苯磺酸、三氟乙酸、十一碳烯酸等)形成。
[0085]“药学上可接受的碱加成盐”是指保持游离酸的生物学有效性和特性的那些盐,其不会在生物学上或其它方面不合需要。这些盐由将无机碱或有机碱添加至游离酸而制备。由无机碱衍生的盐包括但不限于钠、钾、锂、铵、钙、镁、铁、锌、铜、锰、铝盐等。优选的无机盐为铵、钠、钾、钙和镁盐。由有机碱衍生的盐包括但不限于以下的盐:伯胺、仲胺和叔胺、取代的胺(包括天然存在的取代的胺)、环状胺和碱性离子交换树脂诸如氨、异丙胺、三甲胺、二乙胺、三乙胺、三丙胺、二乙醇胺、乙醇胺、丹醇、2-二甲基氨基乙醇、2-二乙基氨基乙醇、二环己基胺、赖氨酸、精氨酸、组氨酸、咖啡因、普鲁卡因、海巴明(hydrabamine)、胆碱、甜菜碱、苯明青霉素、苄星青霉素、乙二胺、葡糖胺、甲基葡糖胺、可可碱、三乙醇胺、氨丁三醇、嘌呤、哌嗪、哌啶、n-乙基哌啶、多胺树脂等的盐。特别优选的有机碱为异丙胺、二乙胺、乙醇
胺、三甲胺、二环己基胺、胆碱和咖啡因。
[0086]
通常结晶产生本发明化合物的溶剂化物。如本文所用的术语“溶剂化物”是指包含一个或多个本发明的化合物的分子与一个或多个溶剂分子的聚集体。所述溶剂可以是水,在该情况下,所述溶剂化物可以是水合物。或者,所述溶剂可以是有机溶剂。因此,本发明化合物可以作为水合物存在,所述水合物包括单水合物、二水合物、半水合物、倍半水合物、三水合物、四水合物等,以及相应的溶剂化的形式。本发明化合物可以是真的溶剂化物,而在其它情况下,本发明化合物可以仅保留外来的水或者是水加上一些外来溶剂的混合物。
[0087]“药物组合物”是指本发明化合物和在用于向哺乳动物(例如,人)递送生物学活性化合物的领域中通常接受的介质的制剂。为此这种介质包括所有药学上可接受的载体、稀释剂或赋形剂。
[0088]“治疗有效量”是指当施用于哺乳动物、优选人时,足以在哺乳动物、优选人或犬中实现如下所定义的治疗的本发明化合物的量。构成“治疗有效量”的本发明化合物或另一种医药剂(例如抗肿瘤剂)的量将根据化合物、病况及其严重程度、施用方式和待治疗的哺乳动物的年龄而变化,但可由本领域普通技术人员根据其自身的知识和本公开内容常规地确定。
[0089]
如本文所用的“治疗(treating)”或“治疗(treatment)”涵盖具有目标疾病或病况的哺乳动物(优选人)的目标疾病或病况的治疗,并且包括:
[0090]
(i)预防疾病或病况在哺乳动物中发生,尤其是当这种哺乳动物倾向于该病况、但还未诊断为有该病况时;
[0091]
(ii)抑制疾病或病况,即,遏制其发展;
[0092]
(iii)减轻疾病或病况,即,引起疾病或病况的减退;或
[0093]
(iv)减轻由疾病或病况导致的症状,即,减轻疼痛,但未解决潜在的疾病或病况。如本文所用的术语“疾病”和“病况”可互换使用,或者可以不同,因为具体的疾病或病况可能不具有已知的成因物质(使得还未研究出病因学),因此还未被认识为疾病,但仅认识到是不合需要的病况或综合征,其中临床医生已鉴定或多或少的一组特定症状。
[0094]
本发明化合物或其药学上可接受的盐可以含有一个或多个不对称中心且因此可产生对映异构体、非对映异构体、及其它立体异构形式,其可根据绝对立体化学而定义为(r)-或(s)-,或针对氨基酸定义为(d)-或(l)-。本发明意在包括所有此类可能的异构体,以及其外消旋及光学纯形式。光学活性的( )和(-)、(r)-和(s)-、或(d)-和(l)-异构体可使用手性合成子或手性试剂来制备,或使用常规技术(例如色谱和分级结晶)来拆分。用于个别对映异构体的制备/分离的常规技术包括由合适的光学纯前体手性合成或使用例如手性高效液相色谱(hplc)拆分外消旋物(或盐或衍生物的外消旋物)。当本文所述的化合物含有烯属双键或其它几何不对称中心时,除非另外说明,否则化合物意欲包括e和z几何异构体两者。同样,还意欲包括所有互变异构形式。
[0095]“立体异构体”是指由相同键所键合的相同原子构成、但具有不同三维结构的化合物,其不可互换。本发明涵盖各种立体异构体及其混合物且包括“对映异构体”,其是指分子是彼此的不可重叠的镜像的两种立体异构体。
[0096]“互变异构体”是指质子从分子的一个原子移位至同一分子的另一原子。本发明包括任何所述化合物的互变异构体。
[0097]
在本发明的范围内还有式(i)的中间体化合物以及上述种类的所有多晶型物和其晶体习性。
附图说明
[0098]
图1是来自评估化合物569在有或没有放射下对mcf7细胞的作用的代表性实验的免疫印迹的图像。
[0099]
图2是示出了在有或没有放射的情况下暴露于媒介物(dmso)或化合物569后检查mcf7细胞活力的克隆源性生存测定的结果的图。
[0100]
图3是示出了化合物569、选择性atm抑制剂(atmi)、选择性dna-pk抑制剂(dna-pki)以及选择性atm抑制剂与选择性dna-pk抑制剂(ai di)的组合在表达野生型p53的hct116细胞或p53表达呈阴性的hct116细胞中诱导tbk1磷酸化的免疫印迹图像。在图3中,p.atm、p.dna-pk、p.tbk1和p.sting分别表示atm、dna-pk、tbk1和sting的磷酸化形式。
[0101]
图4是示出了在fadu头颈部鳞状细胞癌(hnscc)人肿瘤异种移植物中化合物569对dna-pk激酶的放射诱导的自磷酸化和kap1(atm底物)的放射诱导的磷酸化的抑制的免疫印迹。在图4中,pdna-pk和pkap1分别表示dna-pk和kap1的磷酸化形式。
[0102]
图5是示出了在mda-mb-231乳腺癌人肿瘤异种移植物中化合物569对dna-pk激酶的放射诱导的自磷酸化和kap1(atm底物)的放射诱导的磷酸化的抑制的免疫印迹。在图5中,pdna-pk和pkap1分别表示dna-pk和kap1的磷酸化形式。
[0103]
图6示出了用化合物569和/或ir,qd
×
3对fadu皮下人异种移植物小鼠模型的给药。示出了研究中每组随时间的中位相对肿瘤体积。在图6中,“569”意指化合物569,“veh.”意指媒介物,且“rad.”意指放射。
[0104]
图7表示在fadu皮下人异种移植物小鼠模型中用化合物569和/或ir,qd
×
3给药的每组的卡普兰-梅尔(kaplan-meier)无五倍生存率。在图7中,“569”意指化合物569,“veh.”意指媒介物,且“rad.”意指放射。
[0105]
图8示出了用化合物569和/或ir,qd
×
3对mda-mb-231皮下人异种移植物小鼠模型的给药。示出了研究中每组随时间的中位相对肿瘤体积。在图8中,“569”意指化合物569,“veh.”意指媒介物,且“rad.”意指放射。
[0106]
图9表示在mda-mb-231皮下人异种移植物小鼠模型中用化合物569和/或ir,qd
×
3给药的每组的卡普兰-梅尔无五倍生存率数据。在图9中,“569”意指化合物569,“veh.”意指媒介物,且“rad.”意指放射。
[0107]
化合物、组合物和方法
[0108]
本发明提供化合物和组合物,它们可用于例如单独或与放射疗法和/或抗肿瘤疗法组合来治疗肿瘤疾病(例如,癌症,例如本文所述的那些癌症)。化合物可以是式(i)的化合物:
[0109][0110]
或其药学上可接受的盐,
[0111]
其中
[0112]
z是ch、cr3或n;
[0113]
y是chr5或nr6;
[0114]
n是0、1、2或3;
[0115]
r1是
–o–
l
–
n(r7)2或任选取代的四元饱和n-杂环基;
[0116]
r2是c
1-3
烷基;
[0117]
每个r3独立地是卤素或任选取代的c
1-3
烷基;
[0118]
r4是任选取代的烷基;
[0119]
r5是氢、任选取代的c
1-3
烷基或苄氧基;
[0120]
r6是任选取代的c
1-3
烷基;
[0121]
每个r7独立地是h或任选取代的c
1-3
烷基;且
[0122]
l是任选取代的亚乙基。
[0123]
有利地,本发明化合物(例如,化合物568、569或570)可展现针对atm和dna-pk的优良抑制活性。有利地,本发明化合物(例如化合物568、569或570)可展现优良的选择性,如由降低的脱靶活性(例如,mtor抑制、pi3kα/δ抑制和/或herg抑制)所量度的。例如,本发明化合物(例如化合物568、569或570)可具有比atm ic
50
或dna-pk ic
50
大至少10倍(例如,至少20倍)的mtor ic
50
。本发明化合物(例如化合物568、569或570)可具有10nm或更大(例如》100nm)的mtor ic
50
。另外或替代地,本发明化合物(例如化合物568、569或570)可具有比atm ic
50
或dna-pk ic
50
大至少100倍(例如,至少500倍、至少1000倍或至少3000倍)的herg ic
50
,当在相同的化合物浓度下测量时。本发明化合物(例如化合物568、569或570)可具有3μm或更大(例如10μm或更大)的herg ic
50
。
[0124]
有利地,本发明化合物(例如化合物568、569或570)可展现优良的药代动力学特性(例如,c
max
、auc和/或t
1/2
)。
[0125]
在一些实施方案中,所述化合物选自由以下组成的组:
[0126][0127]
本发明化合物的优点在于它们可以抑制atm(共济失调-毛细血管扩张,突变型)和dna-pk激酶。atm(共济失调-毛细血管扩张,突变型)和dna-pk激酶尤其是细胞对dna断裂的反应的重要调节剂并且对这些分子中任一个的抑制都显著增加细胞对电离放射的敏感性。因此,本发明化合物可以是atm和dna-pk作用的有效抑制剂,有或没有放射并且有或没有化学疗法或免疫疗法,以提供治疗肿瘤疾病(例如癌症,例如本文所述的那些癌症)的有效疗法。用本发明化合物治疗患者可以延迟或消除放射疗法对dna损伤的修复。因此,接受本发明化合物的患者可以更好地响应抗肿瘤疗法。有利地,与未接受本发明化合物的患者常规使用相比,接受本发明化合物的患者可以通过从标准剂量的放射疗法增加肿瘤控制或通过从较低剂量的电离放射实现相似水平的肿瘤控制而获得治疗益处。有利地,与未接受本发明化合物的患者所需的剂量相比,较低剂量的电离放射对非癌组织的损害较小。
[0128]
具有分别编码突变型共济失调毛细血管扩张(atm)激酶和dna依赖性蛋白激酶(dna-pk)的atm或prkdc基因的功能丧失突变的人和小鼠对电离放射过敏。atm和dna-pk激酶一起抑制可以有效地使肿瘤细胞对放射或dna损伤剂(例如抗肿瘤剂)敏感。atm和dna-pk激酶双重抑制的功效可能优于单独抑制任一激酶的功效。
[0129]
另外,本发明化合物可有利地展现对其他激酶(atr和mtor)的抑制减小且因此可展现降低的毒性。
[0130]
本发明化合物可使肿瘤细胞对放射和/或抗肿瘤剂敏感。
[0131]
方法
[0132]
另一方面,本发明提供了用于治疗哺乳动物、优选人或犬的肿瘤疾病(例如,癌症)的方法,其中所述方法包括向有此需要的哺乳动物施用治疗有效量的本发明化合物。在一些实施方案中,向接受放射疗法的哺乳动物施用所述化合物。
[0133]
另一方面,本发明提供了用于治疗哺乳动物的肿瘤疾病(例如,癌症)的方法,其中所述方法包括向有此需要的哺乳动物施用治疗有效量的本发明化合物。在一些实施方案中,所述化合物与dna损伤剂组合向哺乳动物施用。dna损伤剂的非限制性实例包括顺铂、奥沙利铂、卡铂、戊柔比星、伊达比星、卡里奇霉素、parp抑制剂。
[0134]
另一方面,本发明提供了药物组合物,其包含本发明化合物和药学上可接受的赋形剂。在一个实施方案中,所述药物组合物包含处于药学上可接受的载剂中且其含量有效治疗动物(优选哺乳动物)的肿瘤疾病的本发明化合物。
[0135]
本发明化合物(当用于组合疗法中时)可以增强其他放射疗法或药物治疗的效力,如果它允许减少其他治疗的剂量的话,这可以降低与其他药物疗法相关的不良事件的发生率和/或严重性。例如,与接受标准全剂量放射疗法而无本发明化合物的患者相比,在接受包括本发明化合物和剂量减少的放射疗法的组合疗法的患者中,放射的副作用(例如,口腔或胃肠粘膜炎、皮炎、肺炎或疲劳)可得以减少(例如,不良事件的发生率可降低了至少1%、5%、10%或20%)。另外,与接受标准全剂量放射疗法而无本发明化合物的患者相比,在接受包括本发明化合物和剂量减少的放射疗法的组合疗法的患者中可得以减少(例如,不良事件的发生率可降低了至少1%、5%、10%或20%)的其他不良事件可以是放射的晚期效应,例如,放射诱导的肺纤维化、心脏损伤、肠梗阻、神经损伤、血管损伤、淋巴水肿、脑坏死或放射诱导的癌症。类似地,当化合物与另一种抗癌药物(例如,本文所述的那些)在组合疗法中给药时,所述组合疗法可能导致相同或甚至增加的肿瘤细胞死亡,即使当另一种抗癌药物的剂量减少时。因此,减少其他抗癌药物的剂量可以降低由其他抗癌药物引起的不良事件的严重性。
[0136]
另一方面,本发明涉及呈其立体异构体、对映异构体、互变异构体或其混合物、或其药学上可接受的盐或溶剂化物形式的如上所述的本发明化合物的用途,或包含药学上可接受的赋形剂和呈其立体异构体、对映异构体、互变异构体或其混合物、或其药学上可接受的盐或溶剂化物形式的如上所述的本发明化合物的药物组合物的用途,它们用于制备供治疗疾病用的药剂。在一些实施方案中,本发明化合物与放射疗法组合施用。在其他实施方案中,本发明化合物与dna损伤剂组合施用。在其他实施方案中,本发明化合物与抗肿瘤免疫治疗剂(例如,易普利单抗、奥法木单抗、纳武单抗、派姆单抗、阿特珠单抗、阿维鲁单抗、德瓦鲁单抗、西米普利单抗(cemiplimab)、奥滨尤妥珠单抗(obinutuzumab)、奥卡妥珠单抗(ocaratuzumab)、替西木单抗、斯巴达珠单抗(spartalizumab)、卡瑞利珠单抗、信迪利单抗(sintilimab)、替雷利珠单抗、特瑞普利单抗(toripalimab)、多塔利单抗(dostarlimab)、维妥珠单抗、incmga00012、amp-224、amp-514、kn035、ck-301、aunp12、ca-170或bms-986189)组合施用。在其他实施方案中,抗肿瘤免疫治疗剂是奥法木单抗、奥滨尤妥珠单抗、
奥卡妥珠单抗或维妥珠单抗。在又其他实施方案中,抗肿瘤免疫治疗剂是纳武单抗、派姆单抗、西米普利单抗、斯巴达珠单抗、卡瑞利珠单抗、信迪利单抗、替雷利珠单抗、特瑞普利单抗、多塔利单抗、incmga00012、amp-224或amp-514。在仍其他实施方案中,抗肿瘤免疫治疗剂是阿特珠单抗、阿维鲁单抗、德瓦鲁单抗、kn035、ck-301、aunp12、ca-170或bms-986189。在某些优选组合疗法实施方案中,本发明化合物与抗肿瘤免疫治疗剂组合施用。
[0137]
本发明方法可用于治疗本文所述的肿瘤疾病。肿瘤疾病可以是例如癌前肿瘤或恶性肿瘤(例如,实体肿瘤或液体肿瘤)。恶性肿瘤通常被称为癌症。在某些实施方案中,肿瘤疾病是癌症。
[0138]
在其他实施方案中,有待使用本文所公开的方法和用途治疗的癌症的实例包括但不限于血液癌症,例如白血病和淋巴瘤。癌症的非限制性实例包括急性骨髓性白血病、急性成淋巴细胞性白血病、急性巨核细胞白血病、早幼粒细胞白血病、红白血病、淋巴细胞性t细胞白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、毛细胞白血病、慢性中性粒细胞白血病、浆细胞瘤、免疫母细胞性大细胞白血病、套细胞白血病、多发性骨髓瘤、恶性淋巴瘤、弥漫性大b细胞淋巴瘤、霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤、成淋巴细胞性t细胞淋巴瘤、伯基特淋巴瘤(burkitt’s lymphoma)和滤泡性淋巴瘤。
[0139]
在又其他实施方案中,有待使用本文所公开的方法和用途治疗的癌症的实例包括但不限于实体肿瘤。实体肿瘤的非限制性实例包括脑癌(例如,星形细胞瘤、胶质瘤、胶质母细胞瘤、髓母细胞瘤或室管膜瘤)、膀胱癌、乳腺癌、中枢神经系统癌、宫颈癌、结肠癌、子宫内膜癌、食道癌、胃肠道间质瘤、胃癌、头颈癌、颊癌、口腔癌、肝细胞癌、肺癌、黑色素瘤、梅克尔细胞癌、间皮瘤、鼻咽癌、神经母细胞瘤、骨肉瘤、卵巢癌、胰腺癌、前列腺癌、肾癌、唾液腺癌、肉瘤、睾丸癌、尿路上皮癌、外阴癌和威尔姆氏瘤。优选地,本发明的方法用于治疗肺癌、头颈癌、胰腺癌、直肠癌、成胶质细胞瘤、肝细胞癌、胆管癌、转移性肝脏病变、黑色素瘤、骨肉瘤、软组织肉瘤、子宫内膜癌、宫颈癌、前列腺癌或梅克尔细胞癌。
[0140]
在仍其他实施方案中,有待使用本文所公开的方法和用途治疗的癌症的实例包括但不限于转移和转移性癌症。例如,本文公开的用于治疗癌症的方法和用途可包括对原发性肿瘤和转移的治疗。
[0141]
在一些实施方案中,本发明的方法可以将受试者的肿瘤大小减小例如至少10%、20%、30%、40%、50%、60%、70%、80%或90%,或可以消除肿瘤(例如,相对于治疗开始时的肿瘤大小或相对于接受安慰剂而不是本发明化合物的参考受试者)。在一些实施方案中,本发明的方法可以将受试者的肿瘤负荷减小例如至少10%、20%、30%、40%、50%、60%、70%、80%或90%,或可以消除肿瘤(例如,相对于治疗开始时的肿瘤负荷或相对于接受安慰剂而不是本发明化合物的参考受试者)。在一些实施方案中,本发明的方法可以将受试者的平均生存时间增加例如至少10%、20%、30%、40%、50%、60%、70%、80%、90%、100%、150%或200%(例如,相对于接受安慰剂而不是本发明化合物的参考受试者)。在一些实施方案中,本发明的方法可以将放射疗法或药物疗法在更长的平均时间内减轻受试者的疼痛或其他症状的能力增强例如至少10%、20%、30%、40%、50%、60%、70%、80%、90%、100%、150%或200%(例如,相对于接受安慰剂而不是本发明化合物的参考受试者)。
[0142]
在一些实施方案中,本文所公开的方法和用途包括在施用放射疗法或dna损伤剂之前用atm和dna-pk双重抑制剂对患者进行预处理。用atm和dna-pk双重抑制剂对患者进行
的预处理可以延迟或消除放射疗法之后dna损伤的修复。
[0143]
放射疗法包括但不限于使用x射线(光子)、来自
60
钴或其他放射性同位素的γ射线、中子、电子、质子、碳离子、氦离子和其他带电粒子进行的外照射放射疗法。放射疗法还包括近距放射疗法和放射性药品,其发射γ射线、α粒子、β粒子、俄歇电子或来自包括以下的同位素的其他类型放射性粒子:
32
磷、
67
铜、
77
溴、
89
锶、
90
钇、
105
铑、
131
碘、
137
铯、
149
钷、
153
钐、
166
钬、
177
镥、
186
铼、
188
铼、
199
金、
211
砹、
213
铋、
223
镭、
225
锕或
227
钍、
192
铱、
67
镓、
103
钯、
125
碘及其他放射性同位素(例如,
192
铱、
125
碘、
137
铯、
103
钯、
32
磷、
90
钇、
67
镓、
211
砹或
223
镭)。放射疗法还包括采用抗体或小分子的放射免疫疗法(rit),这些抗体或小分子缀合至放射性同位素,包括
131
碘、
90
钇、
225
锕、
211
砹、
67
镓、
177
镥、
227
钍及其他放射性同位素。
[0144]
在一些实施方案中,组合疗法包括向患者施用atm和dna-pk抑制剂及抗肿瘤剂,例如顺铂、奥沙利铂、卡铂、拓扑异构酶i抑制剂、拓扑异构酶ii抑制剂、蒽环类、缬柔比星、伊达比星、加利车霉素、parp抑制剂(例如,奥拉帕尼、卢卡帕尼、尼拉帕尼、维利帕尼或他拉唑帕尼),以及本领域技术人员已知的其他抗癌剂。
[0145]
在某些实施方案中,组合疗法包括向患者施用atm和dna-pk抑制剂及抗肿瘤免疫治疗剂,包括但不限于易普利单抗、奥法木单抗、纳武单抗、派姆单抗、阿特珠单抗、阿维鲁单抗、德瓦鲁单抗等。
[0146]
在本文所述的组合疗法中,atm和dna-pk抑制剂可以同时或依次(例如,在其他药物之前或之后)施用于患者。
[0147]
本发明化合物的制备
[0148]
本发明化合物可以使用本领域已知的方法和技术来制备。在实施例中提供了用于合成这些化合物的合适工艺。通常,式(i)的化合物可以根据下述方案制备。还描述了这些反应的起始物料的来源。
[0149]
保护基团可以根据本领域技术人员已知且如本文所述的标准技术在本发明化合物的制备中添加或除去。保护基团的使用详述于greene,t.w.和p.g.m.wuts,greene's protective groups in organic synthesis(2006),第4版,wiley中。保护基团还可以是聚合物树脂,诸如wang树脂或2-氯三苯甲基氯树脂。
[0150]
本领域技术人员还将理解,尽管本发明化合物的此类受保护的衍生物可能不具有原样的药理学活性,但可将它们施用于哺乳动物,且其后在体内代谢,以形成具有药理学活性的本发明化合物。
[0151]
通过用适当的无机或有机碱或酸处理,可以游离碱或酸形式存在的所制备的下文所述的所有化合物可被转化为它们的药学上可接受的盐。可以通过标准技术将下文制备的化合物的盐转化为它们的游离碱或酸形式。应当理解,本发明化合物的所有多晶型物、无定型形式、无水物、水合物、溶剂化物和盐都在本发明的范围内。此外,可通过本领域技术人员已知的方法或通过本文所述的方法分别将所有含有酸或酯基团的本发明化合物转化为相应的酯或酸。
[0152]
这些化合物中的许多化合物的制备的一般表现显示于下面的方案1中。化合物通过偶联分子的各种组分来制备:卤基取代的化合物3(或2’)与硼酸或硼酸酯化合物2(3’)的suzuki偶联。可能需要或可能不需要进一步的反应来提供本发明化合物的合成。本发明的特定化合物的制备显示于以下方案中。
[0153][0154]
方案1
[0155]
在此方法中,可以使用suzuki偶联反应条件将卤素取代转化为芳基取代。此方法的条件公开于许多出版物中,这些出版物已由a.suzuki综述于modern arene chemistry 2002,53-106中的题为“the suzuki reaction with arylboron compounds in arene chemistry”的论文中。在进行此反应中,可以利用suzuki反应中常规的任何合适的条件。通常,suzuki偶联反应在过渡金属催化剂诸如钯催化剂存在下,利用此反应的任何常规有机溶剂和弱无机或有机碱来进行。其中优选的有机溶剂是极性非质子溶剂。任何常规的极性非质子溶剂都可用于制备本发明化合物。合适的溶剂是惯用的,特别是较高沸点的溶剂,例如,二甲氧基乙烷。弱无机碱可以是碳酸盐或碳酸氢盐,诸如碳酸钾或碳酸铯。有机碱可以是胺,诸如三乙胺。
[0156][0157]
方案2
[0158]
具体地,其它螺羟吲哚中间体7如方案2中所示合成。环基或杂环基取代的酯5用强碱诸如但不限于二异丙基氨基锂在低温下在无水溶剂诸如但不限于四氢呋喃中处理,以与起始物料4反应来提供中间体6,所述起始物料4为市售或由本领域技术人员遵循文献描述的方法来制备。中间体6通过还原剂诸如但不限于铁来还原,以得到相应的氨基中间体,所述氨基中间体原位环化以提供羟吲哚化合物7。因此,然后将化合物7用烷基化试剂在碱诸如但不限于碳酸钾或氢化钠存在的情况下在极性溶剂诸如但不限于n,n-二甲基甲酰胺或四氢呋喃中n-烷基化,由此生成螺羟吲哚中间体8。
[0159][0160]
方案3
[0161]
具体地,本发明中的式(i)的化合物可以如方案3中所示合成。市售的5-溴-2-氯-3-硝基-吡啶(9)与亲核试剂xh(10)在强碱诸如但不限于氢化钠存在的情况下反应,以提供中间体11。在钯催化的条件下,可以制备硼酸酯12,所述硼酸酯12然后与螺中间体8反应以提供交叉偶联的产物13。使用还原剂诸如但不限于铁将化合物13中的硝基还原为氨基,以提供中间体14。14与不同磺酰氯(15)的反应提供式(i)的化合物的合成。
[0162][0163]
方案4
[0164]
具体地,本发明中的式(i)的化合物还可以如方案4中所示合成。使用还原剂诸如但不限于铁将化合物11中的硝基还原为氨基,以提供中间体16。16与不同的磺酰氯(15)的反应提供磺酰胺中间体17,其在钯催化下转化为其相应的硼酸酯18。硼酸酯18可以在suzuki反应条件下与卤代化合物8偶联以提供式(i)的化合物。
[0165]
在方案3和方案4中,交叉偶联的化合物也可使用suzuki偶联化学法用具有逆转的卤素和硼酸酯/硼酸取代模式的组分合成,例如,如方案5中所示。
[0166][0167]
方案5
[0168]
具体地,本发明中的式(i)的化合物还可以如方案5中所示合成。卤代化合物8可以在钯催化下转化为其相应的硼酸酯19。硼酸酯19可以在suzuki反应条件下与卤代化合物17偶联以提供式(i)的化合物。
[0169]
在本发明方法的实施中,有效量的任何一种本发明化合物或任何本发明化合物或
其药学上可接受的盐的组合经由任何常用的和可接受的本领域已知的方法单独或组合地施用。因此,所述化合物或组合物可以经口(例如颊腔)、舌下、胃肠外(例如肌内、静脉内或皮下)、直肠(例如通过栓剂或洗剂)、经皮(例如皮肤电穿孔)或通过吸入(例如通过气溶胶)并且以形状或固体、液体或气体给药(包括片剂和悬浮液)来施用。可以单一单位剂型用连续疗法或以单剂量疗法任意施用。所述治疗组合物还可以呈与亲脂性盐诸如双羟萘酸结合的油乳液或分散体的形式,或者呈用于皮下或肌内施用的可生物降解的缓释组合物的形式。
[0170]
用于制备其组合物的有用的药物载体可以是固体、液体或气体;因此,所述组合物可以采取片剂、丸剂、胶囊、栓剂、粉剂、肠溶包衣或其它保护制剂(例如在离子交换树脂上结合或在脂质-蛋白质囊泡中包装)、缓释制剂、溶液、悬浮液、酏剂、气溶胶等形式。所述载体可以选自各种油,包括石油、动物、植物或合成来源的油,例如花生油、大豆油、矿物油、芝麻油等。水、盐水、右旋糖水溶液和二醇类是优选的液体载体,特别是(当与血液等渗时)用于可注射溶液。例如,用于静脉内施用的制剂包含活性成分的无菌水溶液,其通过将固体活性成分溶解在水中以制备水溶液并使溶液无菌而制备。合适的药物赋形剂包括淀粉、纤维素、滑石、葡萄糖、乳糖、明胶、麦芽、稻米、面粉、白垩、二氧化硅、硬脂酸镁、硬脂酸钠、单硬脂酸甘油酯、氯化钠、脱脂奶粉、甘油、丙二醇、水、乙醇等。所述组合物可以接受常规药物添加剂,诸如防腐剂、稳定剂、润湿剂或乳化剂、用于调节渗透压的盐、缓冲剂等。合适的药物载体及其制剂描述于e.w.martin的remington's pharmaceutical sciences中。无论如何,此类组合物将含有有效量的活性化合物以及合适的载体,以便制备用于适当地施用于接受者的合适剂型。
[0171]
本发明化合物的剂量取决于许多因素,诸如例如施用方式、患者的年龄和体重、以及待治疗的患者的病况,并且最终将由主治医师或兽医决定。由主治医师或兽医确定的活性化合物的这种量在本文中以及在权利要求中称为“有效量”。
[0172]
现在将在下面的实施例中进一步描述本发明,所述实施例仅意欲作为举例说明且不限制本发明的范围。
[0173]
实施例
[0174]
试剂购自aldrich,sigma,tci(shanghai)development,chembon pharmaceutical co.,ltd,zhangjiagang aimate huaxue youxiangongsi,changzhou qinuo biotech co.ltd和shanghai weiyuan fine fluorine technology development co.,ltd或如下所示的其它供应商并且不经进一步纯化即使用。使用微波放射用于加热的反应使用biotage initiator 进行。通过本领域技术人员已知的方法诸如硅胶快速柱色谱的洗脱进行多毫克至多克规模的纯化;在一些情况下,也通过使用用biotage combiflash系统洗脱的处理预填充硅胶柱(welch/agela)实现制备型快速柱色谱纯化。
[0175]
出于判断化合物身份和纯度的目的,通常使用分析型lc-ms(液相色谱/质谱)系统,其由具有阳离子检测模式的电喷雾电离的waters zq
tm
平台与具有自动进样器的agilent 1100系列hplc组成。该柱通常是water xterra ms c18,3.0
×
50mm,5μm。流速为1ml/min,且注射体积为10μl。uv检测在范围210-400nm内。流动相由溶剂a(水 0.06%tfa)和溶剂b(乙腈 0.05%tfa)组成,梯度为100%溶剂a持续0.7min,经3.75min变成100%溶剂b,保持1.1min,随后经0.2min回到100%溶剂a。
氯-7-氟-3-硝基喹啉(1.50g,4.91mmol)。在环境温度下再搅拌1小时之后,将反应通过饱和氯化铵水溶液(60.0ml)猝灭并用水(120ml)稀释。将所得混合物用乙酸乙酯(3x60.0ml)萃取。将合并的有机层用盐水(2x50.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~2%乙酸乙酯/石油醚洗脱,得到呈无色固体状的标题化合物(240mg,13%):1h nmr(400mhz,cdcl3)δ9.14(s,1h),8.20(d,j=7.2hz,1h),7.90(d,j=8.8hz,1h),3.84(s,3h),3.12-2.99(m,1h),2.58-2.48(m,3h),1.91-1.83(m,1h),1.45-1.27(m,1h);ms:[(m 1)]
=383.17,385.17。
[0201][0202]
8'-溴-7'-氟螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:将1-(6-溴-7-氟-3-硝基喹啉-4-基)环丁烷-1-甲酸甲酯(240mg,0.63mmol)和铁粉(350mg,6.26mmol)于乙酸(10.0ml)中的混合物在环境温度下搅拌18小时。将所得混合物过滤,并将滤饼用乙酸乙酯(5x100ml)洗涤。在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~2%甲醇/二氯甲烷洗脱,得到呈淡黄色固体状的标题化合物(100mg,50%):1h nmr(400mhz,dmso-d6)δ10.75(s,1h),8.68(s,1h),8.52(d,j=7.5hz,1h),7.98(d,j=10.1hz,1h),2.90-2.75(m,2h),2.50-2.37(m,4h);ms:[(m 1)]
=321.15,323.15。
[0203][0204]
8'-溴-7'-氟-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:在氮气气氛下,在0℃下将8-溴-7-氟-2,3-二氢螺[环丁烷-1,1-吡咯并[2,3-c]喹啉]-2-酮(100mg,0.31mmol)于n,n-二甲基甲酰胺(10.0ml)中的溶液用氢化钠(19.9mg,0.50mmol,60%,分散于矿物油中)处理30min,接着添加碘甲烷(66.3mg,0.47mmol)。在环境温度下再搅拌40分钟之后,将反应通过饱和氯化铵水溶液(10.0ml)猝灭。将所得混合物用水(100ml)稀释并用乙酸乙酯(3x30.0ml)萃取。将合并的有机层用盐水(2x20.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过prep-tlc(dcm/meoh=20/1,v:v)纯化以得到呈无色固体状的标题化合物(102mg,98%):1h nmr(400mhz,cd3od)δ8.78(s,1h),8.61(d,j=7.4hz,1h),7.85(d,j=9.8hz,1h),3.36(s,3h),2.94-2.85(m,2h),2.72-2.61(m,3h),2.56-2.48(m,1h);ms:[(m 1)]
=335.00,337.00。
[0205][0206]
n-(2-羟乙基)-n-(丙烷-2-基)氨基甲酸叔丁酯:在0℃下向2-[(丙烷-2-基)氨基]乙-1-醇(40.0g,388mmol)于甲醇(300ml)中的溶液中逐滴添加二碳酸二叔丁酯(127g,586mmol)。在环境温度下搅拌所得混合物2小时,并在减压下浓缩。将残余物通过硅胶柱色谱纯化,用0%~4%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈无色
油状的标题化合物(65.0g,82%):1h nmr(400mhz,cdcl3)δ4.17(s,1h),3.71(t,j=5.4hz,2h),3.30(t,j=5.4hz,2h),1.47(s,9h),1.12(d,j=6.8hz,6h)。
[0207][0208]
n-[2-[(5-溴-3-硝基吡啶-2-基)氧基]乙基]-n-(丙烷-2-基)氨基甲酸叔丁酯:在氮气气氛下,在0℃下将n-(2-羟乙基)-n-(丙烷-2-基)氨基甲酸叔丁酯(15.4g,75.8mmol)于无水四氢呋喃(250ml)中的溶液用氢化钠(3.30g,82.1mmol,60%w/w,分散于矿物油中)处理1小时,接着在0℃下经2分钟添加5-溴-2-氯-3-硝基吡啶(15.0g,63.2mmol)。在25℃下再搅拌2小时之后,将反应通过饱和氯化铵水溶液(50.0ml)猝灭并用水(500ml)稀释。将水层用乙酸乙酯(3x150ml)萃取。将合并的有机层用盐水(2x100ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~18%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈淡黄色油状的标题化合物(18.0g,71%):1h nmr(400mhz,cdcl3)δ8.42(d,j=2.4hz,1h),8.37(d,j=2.4hz,1h),4.57(t,j=6.3hz,2h),4.32(s,1h),3.51(t,j=6.3hz,2h),1.47(s,9h),1.15(d,j=6.9hz,6h);ms:[(m 1)]
=404.00,406.00。
[0209][0210]
n-[2-[(3-氨基-5-溴吡啶-2-基)氧基]乙基]-n-(丙烷-2-基)氨基甲酸叔丁酯:在环境温度下,向n-[2-[(5-溴-3-硝基吡啶-2-基)氧基]乙基]-n-(丙烷-2-基)氨基甲酸叔丁酯(15.0g,37.1mmol)于乙酸(150ml)中的溶液中添加铁粉(20.7g,371mmol)。在环境温度下再搅拌1小时之后,将所得混合物过滤,并将滤饼用四氢呋喃(4x100ml)洗涤。在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用20%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(12.0g,86%):1h nmr(400mhz,cd3od)δ7.40(d,j=2.2hz,1h),7.04(d,j=2.2hz,1h),4.40(t,j=6.3hz,2h),4.25-3.99(m,1h),3.52(t,j=6.3hz,2h),1.46(s,9h),1.17(d,j=6.8hz,6h);ms:[(m 1)]
=374.10,376.10。
[0211][0212]
(2-((5-溴-3-(甲基磺酰氨基)吡啶-2-基)氧基)乙基)(异丙基)氨基甲酸叔丁酯:在环境温度下,向n-[2-[(3-氨基-5-溴吡啶-2-基)氧基]乙基]-n-(丙烷-2-基)氨基甲酸叔丁酯(18.8g,50.3mmol)于吡啶(400ml)中的溶液中逐滴添加甲磺酰氯(8.63g,75.4mmol)。在环境温度下搅拌6小时之后,在减压下浓缩所得混合物。将残余物通过硅胶柱色谱纯化,用3%~25%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(16.3g,72%):1h nmr(400mhz,dmso-d6)δ9.41(s,1h),8.05(d,j=2.2hz,
1h),7.79(d,j=2.2hz,1h),4.35(t,j=6.3hz,2h),4.15(s,1h),3.43(t,j=6.3hz,2h),3.11(s,3h),1.39(s,9h),1.09(d,j=6.8hz,6h);ms:[(m 1)]
=452.00,454.00。
[0213][0214]
(2-((5-溴-3-(乙基磺酰氨基)吡啶-2-基)氧基)乙基)(异丙基)氨基甲酸叔丁酯:在氮气气氛下,在环境温度下向(2-((3-氨基-5-溴吡啶-2-基)氧基)乙基)(异丙基)氨基甲酸叔丁酯(5.00g,13.4mmol)于吡啶(120ml)中的搅拌溶液中逐滴添加乙磺酰氯(5.15g,40.1mmol)。在氮气气氛下,在环境温度下搅拌6小时之后,在减压下浓缩所得混合物。将残余物通过硅胶柱色谱纯化,用3%~25%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈淡黄色固体状的标题化合物(3.78g,61%):1h nmr(400mhz,cd3od)δ7.92(s,1h),7.87(s,1h),4.48(t,j=6.6hz,2h),4.25-4.20(m,1h),3.54(t,j=6.6hz,2h),3.13(t,j=7.3hz,2h),1.49(s,9h),1.35(t,j=7.4hz,3h),1.19(d,j=6.8hz,6h);ms:[(m 1)]
=466.10,468.10。
[0215][0216]
n-[2-[(5-溴-3-碘吡啶-2-基)氧基]乙基]-n-异丙基氨基甲酸叔丁酯:在0℃下向5-溴-3-碘吡啶-2-醇(10.0g,33.3mmol)于无水四氢呋喃(300ml)中的溶液中逐滴添加三苯基膦(11.4g,43.3mmol)、n-(2-羟乙基)-n-异丙基氨基甲酸叔丁酯(8.80g,43.3mmol)和偶氮二甲酸二异丙酯(8.80g,43.4mmol)。在环境温度下,在氮气气氛下再搅拌所得混合物16小时。在减压下浓缩所得混合物。将残余物通过硅胶柱色谱纯化,用2%~10%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈无色油状的标题化合物(14.0g,87%):1h nmr(400mhz,cdcl3)δ8.14(s,2h),4.43(t,j=6.4hz,2h),4.38-3.96(m,1h),3.50(t,j=6.4hz,2h),1.49(s,9h),1.19(d,j=6.8hz,6h);ms:[(m 1)]
=484.95,486.95。
[0217][0218]
(2-((5-溴-3-((1-甲基乙基)磺酰氨基)吡啶-2-基)氧基)乙基)(异丙基)氨基甲酸叔丁酯:在环境温度下,向(2-((5-溴-3-碘吡啶-2-基)氧基)乙基)(异丙基)氨基甲酸叔丁酯(5.00g,10.3mmol)、4,5-双(二苯基膦)-9,9-二甲基呫吨(1.80g,3.10mmol)和丙烷-2-磺酰胺(1.50g,12.4mmol)于甲苯(125ml)中的混合物中添加磷酸三钾(10.9g,51.5mmol)和三(二亚苄基丙酮)二钯-氯仿加合物(1.10g,1.10mmol)。在氩气气氛下,在100℃下搅拌所得混合物48小时。冷却至环境温度之后,将所得混合物过滤。将滤饼用乙酸乙酯(3x20ml)洗
涤。在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~20%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(1.90g,39%):1h nmr(400mhz,cdcl3)δ7.94(d,j=2.1hz,1h),7.89(d,j=2.2hz,1h),4.44(t,j=6.3hz,2h),4.11(s,1h),3.48(t,j=6.3hz,2h),3.24-3.30(m,1h),1.48(s,9h),1.41(d,j=6.8hz,6h),1.14(d,j=6.8hz,6h);ms:[(m 1)]
=480.20,482.20。
[0219][0220]
n-(5-溴-2-[2-[(丙烷-2-基)氨基]乙氧基]吡啶-3-基)甲磺酰胺:在环境温度下,将向n-[2-[(5-溴-3-甲磺酰氨基吡啶-2-基)氧基]乙基]-n-(丙烷-2-基)氨基甲酸叔丁酯(3.00g,6.63mmol)于二氯甲烷(5.00ml)中的溶液用氯化氢(20.0ml,4m,于1,4-二噁烷中)处理40min。在减压下浓缩所得混合物。将残余物用饱和碳酸氢钠水溶液(30.0ml)碱化至ph=8。将所得混合物用乙酸乙酯(6x200ml)萃取。将合并的有机层经无水硫酸钠干燥并过滤。在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~10%甲醇/二氯甲烷洗脱。将所需级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(1.20g,50%):1h nmr(400mhz,dmso-d6)δ7.68(d,j=2.3hz,1h),7.61(d,j=2.3hz,1h),5.75(s,1h),4.36(t,j=5.2hz,2h),3.19-3.12(m,1h),3.07(t,j=5.1hz,2h),2.84(s,3h),1.15(d,j=6.4hz,6h);ms:[(m 1)]
=352.10,354.10。
[0221]
根据以上所述的程序制备下列中间体:
[0222][0223][0224]
n-(2-(2-(异丙氨基)乙氧基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-3-基)甲磺酰胺:在环境温度下,向n-[2-[(5-溴-3-甲磺酰氨基吡啶-2-基)氧基]乙基]-n-(丙烷-2-基)氨基甲酸叔丁酯(2.00g,5.68mmol)和双(频哪醇合)二硼(2.88g,11.4mmol)于1,4-二噁烷(50.0ml)中的溶液中添加乙酸钾(2.23g,22.7mmol)和双(二苯基膦)二茂铁]二氯钯(ii)二氯甲烷加合物(0.46g,0.57mmol)。在氮气气氛下,在90℃下搅拌2小时之后,在减压下浓缩所得混合物。用二氯甲烷(100ml)稀释所得混合物。过滤之后,将滤
饼用二氯甲烷(3x10.0ml)洗涤。在减压下浓缩滤液。将残余物通过反相快速色谱以下列条件下纯化:柱:spherical c18,20~40μm,330g;流动相a:水(加上10mm nh4hco3),流动相b:乙腈;流率:65ml/min;梯度(b%):5%~22%,6min;22%~40%,20min;40%~95%;2min;95%,5min;检测器:uv 254nm;rt:15min。将含有所需产物的级分收集并在减压下浓缩以得到呈灰白色固体状的标题化合物(1.57g,87%):1h nmr(400mhz,cdcl3)δ8.27(d,j=1.7hz,1h),8.06(d,j=1.7hz,1h),4.51(t,j=5.2hz,2h),3.05-2.99(m,5h),2.95-2.84(m,1h),1.33(s,12h),1.11(d,j=6.3hz,6h);ms:[(m 1)]
=400.30。
[0225]
化合物568
[0226][0227]
n-(5-(7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)丙烷-2-磺酰胺:在环境温度下,向n-[5-溴-2-[2-(异丙氨基)乙氧基]吡啶-3-基]丙烷-2-磺酰胺(2.00g,5.26mmol)于1,4-二噁烷(50.0ml)中的溶液中添加双(频哪醇合)二硼(4.01g,15.8mmol)、乙酸钾(2.06g,21.1mmol)和双(二苯基膦)二茂铁]二氯钯(ii)二氯甲烷加合物(0.34g,0.42mmol)。在氮气气氛下,在90℃下搅拌所得混合物3小时。将所得混合物冷却至环境温度,接着添加8-溴-7-氟-3-甲基螺[环丁烷-1,1-吡咯并[2,3-c]喹啉]-2-酮(1.26g,3.76mmol)、水(12.5ml)、碳酸钠(0.80g,7.55mmol)和四(三苯基膦)钯(0)(0.43g,0.38mmol)。在氮气气氛下,在85℃下搅拌所得混合物6小时。在冷却至环境温度之后,在减压下浓缩所得混合物。将残余物通过反相快速色谱以下列条件纯化:柱:spherical c18,20~40μm,120g;流动相a:水(加上10mm nh4hco3);流动相b:乙腈;流率:45ml/min;梯度(b%):5%,2min;5%~25%,8min;25%~39%,9min;39%,10min;39%~95%;3min;95%,2min;检测器:uv 254nm;rt:21min。将含有所需产物的级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(1.66g,80%):1h nmr(400mhz,dmso-d6)δ8.88(s,1h),8.35-8.31(m,2h),8.07(s,1h),7.99(s,1h),7.96(s,1h),4.55(br,1h),4.44(t,j=5.2hz,2h),3.35-3.30(m,4h),2.93-2.79(m,5h),2.55-2.38(m,4h),1.29(d,j=7.2hz,6h),1.02(d,j=6.0hz,6h);ms:[(m 1)]
=556.30。
[0228]
[0229]
n-(5-(7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)丙烷-2-磺酰胺氢氯酸.将n-(5-[7-氟-3-甲基-2-氧代螺[环丁烷-1,1-吡咯并[2,3-c]喹啉]-8-基]-2-[2-(异丙氨基)乙氧基]吡啶-3-基)丙烷-2-磺酰胺(1.66g,2.99mmol)于稀盐酸水溶液(392ml,3.14mmol,0.008m)和乙腈(79.0ml)中的溶液冻干以得到呈黄色固体状的标题化合物(1.76g,100%):1h nmr(400mhz,dmso-d6)δ9.45(s,1h),9.02(br,2h),8.90(s,1h),8.40(s,1h),8.32(d,j=8.4hz,1h),8.16(s,1h),8.00(d,j=8.4hz,1h),4.65(t,j=4.4hz,2h),3.54-3.40(m,3h),3.31(s,3h),2.91(t,j=11.2hz,2h),2.55-2.45(m,5h),1.34-1.30(m,12h);ms:[(m 1)]
=556.30。
[0230]
以类似的方式或用中间体cc108和cc110实现钯(ii)偶联以得到下列化合物实施例569和实施例570。
[0231]
[0232][0233]
化合物571
[0234][0235]
3-(6-溴-7-氟-3-硝基喹啉-4-基)氮杂环丁烷-1,3-二甲酸1-(叔丁酯)3-甲酯:在-78℃下向新近制备的二异丙基氨基锂(137mmol)于无水四氢呋喃(110ml)中的溶液中添加氮杂环丁烷-1,3-二甲酸1-叔丁酯3-甲酯(29.3g,137mmol)于四氢呋喃(100ml)中的溶液。搅拌1小时之后,经20min将6-溴-4-氯-7-氟-3-硝基喹啉(32.0g,105mmol)于四氢呋喃(100ml)中的溶液添加到反应混合物中。将所得混合物缓慢温至0℃并持续2小时。将反应通过饱和氯化铵水溶液(20.0ml)猝灭并用水(800ml)稀释。将所得混合物用乙酸乙酯(3x150ml)萃取。将合并的有机层用盐水(2x100ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用5%~20%乙酸乙酯/石油醚洗脱,得到呈淡黄色固体状的标题化合物(25.0g,49%):1h nmr(400mhz,dmso-d6)δ9.35(s,1h),8.21(d,j=9.3hz,1h),8.14(d,j=7.1hz,1h),4.18-4.11(m,2h),3.87-3.74(m,2h),3.66(s,3h),1.37(s,9h);ms:[(m 1)]
=484.20,486.20。
[0236][0237]
8'-溴-7'-氟-2'-氧代-2',3'-二氢螺[氮杂环丁烷-3,1'-吡咯并[2,3-c]喹啉]-1-甲酸叔丁酯:
[0238]
在环境温度下,向3-(6-溴-7-氟-3-硝基喹啉-4-基)氮杂环丁烷-1,3-二甲酸1-叔丁酯3-甲酯(12.0g,24.8mmol)于乙酸(300ml)中的溶液中添加铁粉(9.69g,174mmol)。在环境温度下搅拌3小时之后,在减压下浓缩所得混合物。将残余物溶于水(200ml)中并用乙酸乙酯(4x100ml)萃取。将合并的有机层用盐水(2x100ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~5%甲醇/二氯甲烷洗脱,得到呈无色固体状的标题化合物(10.4g,99%):1h nmr(400mhz,dmso-d6)δ11.02(br,1h),8.71(s,1h),8.24(d,j=7.3hz,1h),8.03(d,j=10.1hz,1h),4.29(d,j=9.0hz,2h),4.21(d,j=9.0hz,2h),1.49(s,9h);ms:[(m 1)]
=422.20,424.20。
[0239][0240]
8'-溴-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[氮杂环丁烷-3,1'-吡咯并[2,3-c]喹啉]-1-甲酸叔丁酯:在氮气气氛下,在0℃下向8-溴-7-氟-2-氧代-2,3-二氢螺[氮杂环丁烷-3,1-吡咯并[2,3-c]喹啉]-1-甲酸叔丁酯(4.22g,9.99mmol)于n,n-二甲基甲酰胺
(100ml)中的搅拌溶液中添加氢化钠(0.52g,13.0mmol,60%,分散于矿物油中)。在环境温度下搅拌所得混合物1小时,接着添加碘甲烷(1.70g,12.0mmol)。在环境温度下再搅拌1小时之后,将反应物通过饱和氯化铵水溶液(20.0ml)猝灭并用水(1.00l)稀释。将沉淀的固体通过过滤收集,用水(3x30.0ml)和己烷(2x30.0ml)洗涤。将所得固体在红外光下干燥以得到呈淡黄色固体状的标题化合物(3.93g,90%):1h nmr(400mhz,cdcl3)δ8.71(s,1h),8.47(d,j=7.0hz,1h),7.94(d,j=9.1hz,1h),4.54(d,j=9.1hz,2h),4.31(d,j=9.0hz,2h),3.40(s,3h),1.56(s,9h);ms:[(m 1)]
=436.15,438.15。
[0241][0242]
8-溴-7-氟-3-甲基-2,3-二氢螺[氮杂环丁烷-3,1-吡咯并[2,3-c]喹啉]-2-酮:将8-溴-7-氟-3-甲基-2-氧代-2,3-二氢螺[氮杂环丁烷-3,1-吡咯并[2,3-c]喹啉]-1-甲酸叔丁酯(3.93g,9.01mmol)和三氟乙酸(20.0ml)于二氯甲烷(100ml)中的溶液在环境温度下搅拌5小时。在减压下浓缩所得混合物。将残余物用饱和碳酸氢钠水溶液碱化至ph=8。将所得混合物用乙酸乙酯(6x300ml)萃取。将合并的有机层用盐水(2x300ml)洗涤并经无水硫酸钠干燥。过滤之后,将滤液在减压下浓缩以得到呈淡黄色固体状的标题化合物(3.00g,99%):1h nmr(400mhz,dmso-d6)δ9.56(d,j=7.7hz,1h),8.92(s,1h),8.02(d,j=10.2hz,1h),4.18(d,j=7.5hz,2h),3.59(d,j=7.5hz,2h),3.29(s,3h);ms:[(m 1)]
=335.95,337.95。
[0243][0244]
8'-溴-7'-氟-1-异丙基-3'-甲基螺[氮杂环丁烷-3,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:在环境温度下,向8'-溴-7'-氟-3'-甲基螺[氮杂环丁烷-3,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮(100mg,0.30mmol)和丙酮(4.00ml)于乙醇(8.00ml)中的搅拌溶液中添加氰基硼氢化钠(94.0mg,1.50mmol)。在氮气气氛下,在50℃下搅拌所得混合物3小时。将所得混合物通过反相快速色谱用下列条件纯化:柱:spherical c18,20~40μm,120g;流动相a:水(加上10mm nh4hco3);流动相b:乙腈;流率:50ml/min;梯度(b):5%~20%,6min;20%~50%,30min;50%~95%,5min;95%,5min;检测器:uv 254nm。将所需级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(85.0mg,77%):1h nmr:1h nmr(400mhz,dmso-d6)δ9.84(d,j=8.0hz,1h),8.93(s,1h),8.00(d,j=10.2hz,1h),3.69(d,j=7.6hz,2h),3.46(d,j=7.3hz,2h),3.30(s,3h),2.63-2.56(m,1h),1.01(d,j=6.1hz,6h);ms:[(m 1)]
=378.10,380.10。
[0245][0246]
n-[5-[7-氟-3-甲基-2-氧代-1-(丙烷-2-基)-2,3-二氢螺[氮杂环丁烷-3,1-吡咯并[2,3-c]喹啉]-8-基]-2-[2-[(丙烷-2-基)氨基]乙氧基]吡啶-3-基]甲磺酰胺:向8-溴-7-氟-3-甲基-1-(丙烷-2-基)-2,3-二氢螺[氮杂环丁烷-3,1-吡咯并[2,3-c]喹啉]-2-酮(80.0mg,0.21mmol)和n-(2-[2-[(丙烷-2-基)氨基]乙氧基]-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-3-基)甲磺酰胺(169mg,0.42mmol)于水(2.00ml)和1,4-二噁烷(10.00ml)中的溶液中添加碳酸钠(33.6mg,0.32mmol)和四(三苯基膦)钯(0)(24.3mg,0.021mmol)。在氮气气氛下,在80℃下搅拌2小时之后,在减压下浓缩所得混合物。将残余物通过prep-tlc(dcm/meoh=10/1,v/v)纯化以得到粗产物,将该粗产物通过反相快速色谱用下列条件进一步纯化:柱:spherical c18,20~40μm,120g;流动相a:水(加上10mm nh4hco3),流动相b:乙腈;流率:45ml/min;梯度(b%):5%,2min;5%~24%,5min;24%~34%,9min;34%,8min;34%~95%;3min;95%,2min;检测器:uv 254nm;rt:21min。将所需级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(65.0mg,54%):1h nmr(400mhz,dmso-d6)δ9.66(d,j=8.9hz,1h),8.89(s,1h),8.25(t,j=1.9hz,1h),7.99-7.91(m,2h),4.42(t,j=5.4hz,2h),3.76(d,j=7.2hz,2h),3.47(d,j=7.3hz,2h),3.31(s,3h),3.02(s,3h),2.96(t,j=5.4hz,2h),2.92-2.84(m,1h),2.62-2.53(m,1h),1.05(d,j=6.3hz,6h),0.97(d,j=6.1hz,6h);ms:[(m 1)]
=571.25。
[0247]
化合物572
[0248][0249]
1-(6-溴-7-氟-3-硝基喹啉-4-基)-3-甲氧基环丁烷-1-甲酸甲酯:在-78℃下向新近制备的二异丙基氨基锂(10.6mmol)于无水四氢呋喃(100ml)中的溶液中添加3-甲氧基环丁烷-1-甲酸甲酯(1.53g,10.6mmol)。再搅拌1小时之后,经5min添加6-溴-4-氯-7-氟-3-硝基喹啉(2.50g,8.18mmol)于四氢呋喃(5.00ml)中的溶液。将所得混合物缓慢温至0℃,然后通过饱和氯化铵水溶液(100ml)猝灭。将所得混合物用水(1.00l)稀释并将有机层分离。将水层用乙酸乙酯(3x200ml)萃取。将合并的有机层用盐水(2x200ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用5%~50%乙酸乙酯/石油醚洗脱,得到呈褐色糖浆状的标题化合物(720mg,21%):1h nmr(400mhz,dmso-d6)δ9.25(s,1h),8.31(d,j=7.3hz,1h),8.17(d,j=9.3hz,1h),4.16-4.13(m,1h),3.71(s,3h),3.11(s,3h),2.47-2.38(m,2h),2.00-1.89(m,2h);ms:[(m 1)]
=413.20,415.20。
[0250][0251]
8'-溴-7'-氟-3-甲氧基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:向1-(6-溴-7-氟-3-硝基喹啉-4-基)-3-甲氧基环丁烷-1-甲酸甲酯(720mg,1.74mmol)于乙酸(10.0ml)中的溶液中添加铁粉(681mg,12.2mmol)。在环境温度下搅拌所得混合物3小时。将所得混合物用水(150ml)稀释并用乙酸乙酯(4x50.0ml)萃取。将合并的有机层用盐水(2x50.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用5%~30%乙酸乙酯/石油醚洗脱,得到呈淡黄色固体状的标题化合物(550mg,89%):1h nmr(400mhz,cdcl3)δ8.93(s,0.45h),8.88(d,j=7.2hz,0.55h),8.84(s,1h),8.78(s,0.55h),8.27(d,j=7.0hz,0.45h)7.92(d,j=9.3hz,1h),4.71-4.63(m,0.45h),4.57-4.49(m,0.55h),3.48(d,j=6.6hz,3h),3.07-2.83(m,4h);ms:[(m 1)]
=351.00,353.00。
[0252][0253]
8'-溴-7'-氟-3-甲氧基-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:在0℃下将8-溴-7-氟-3-甲氧基-2,3-二氢螺[环丁烷-1,1-吡咯并[2,3-c]喹啉]-2-酮(550mg,1.56mmol)于n,n-二甲基甲酰胺(10.0ml)中的溶液用氢化钠(81.4mg,2.04mmol,60%,分散于矿物油中)处理0.5小时,然后在0℃下经2min逐滴添加碘甲烷(265mg,1.87mmol)。在环境温度下再搅拌1小时之后,将反应通过饱和氯化铵水溶液(20.0ml)猝灭并用水(150ml)稀释。将所得混合物用乙酸乙酯(3x50.0ml)萃取。将合并的有机层用盐水(2x30.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用5%~10%乙酸乙酯/石油醚洗脱,得到呈黄色固体状的标题化合物(500mg,87%):ms:[(m 1)]
=365.10,367.10。
[0254][0255]
8'-溴-7'-氟-3-羟基-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:在氮气气氛下,在-78℃下向8'-溴-7'-氟-3-甲氧基-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮(900mg,2.46mmol)于二氯甲烷(20.0ml)中的搅拌溶液中逐滴添加三溴化硼(24.6ml,24.6mmol,1m,于二氯甲烷中)。将所得混合物缓慢升温至环境温度。在氮气气氛下,在环境温度下搅拌所得混合物2小时。将混合物用饱和碳酸氢钠水溶液中和。
将水层用乙酸乙酯(3x100ml)萃取。将合并的有机层用盐水(3x100ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~5%甲醇/二氯甲烷洗脱。将所需级分收集并在减压下浓缩以得到呈无色固体状的标题化合物(530mg,62%):1h nmr(400mhz,dmso-d6)δ8.96-8.89(m,1.6h),8.32(d,j=7.4hz,0.4h),8.05-7.98(m,1h),5.99(d,j=6.5hz,0.6h),5.73(d,j=5.7hz,0.4h),4.97-4.87(m,0.4h),4.75-4.65(m,0.6h),3.31(s,1.2h),3.29(s,1.8h),2.98-2.87(m,0.8h),2.81-2.67(m,2.4h),2.65-2.55(m,0.8h);ms:[(m 1)]
=351.00,353.00。
[0256][0257]
3-(苄氧基)-8'-溴-7'-氟-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:
[0258]
在氮气气氛下,在0℃下向8'-溴-7'-氟-3-羟基-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮(100mg,0.29mmol)于n,n-二甲基甲酰胺(5.00ml)中的溶液中添加氢化钠(13.7mg,0.35mmol,60%,分散于矿物油中)。将所得混合物在25℃下搅拌1小时,然后在0℃下添加苄基溴(97.4mg,0.57mmol)。在25℃下再搅拌1小时之后,将反应通过饱和氯化铵水溶液(10.0ml)猝灭。将所得混合物用水(100ml)稀释并用乙酸乙酯(3x50.0ml)萃取。将合并的有机层用盐水(2x50.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过prep-tlc(dcm/meoh=25/1,v/v)纯化以得到呈黄色固体状的标题化合物(64.0mg,51%):1h nmr(400mhz,dmso-d6)δ8.92(d,j=3.6hz,1h),8.79(d,j=7.6hz,0.5h),8.25(d,j=7.2hz,0.5h),8.03(d,j=10.1hz,1h),7.50(d,j=7.0hz,1h),7.46-7.37(m,3h),7.37-7.30(m,1h),4.87-4.79(m,0.5h),4.62-4.50(m,2.5h),3.30(d,j=5.7hz,3h),2.98-2.90(m,1h),2.80(d,j=6.7hz,2h),2.72-2.64(m,1h);ms:[(m 1)]
=441.00,443.00。
[0259][0260]
n-(5-(3-(苄氧基)-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺:向3-(苄氧基)-8'-溴-7'-氟-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮(64.0mg,0.15mmol)和n-[2-[2-(异丙氨基)乙氧基]-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-3-基]甲磺酰胺(69.5mg,0.17mmol)于水(1.00ml)和1,4-二噁烷(4.00ml)中的溶液中添加碳酸钠(15.4mg,0.15mmol)和四(三苯基膦)钯(0)(33.5mg,0.029mmol)。在氮气
气氛下,在80℃下搅拌2小时之后,在减压下浓缩所得混合物。将残余物通过prep-tlc(dcm/meoh=10/1,v/v)纯化以得到呈淡黄色固体状的标题化合物(55.0mg,60%):ms:[(m 1)]
=634.55。
[0261][0262]
顺式-n-(5-(3-(苄氧基)-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(实施例572)和反式-n-(5-(3-(苄氧基)-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(实施例567)。将上述n-(5-(3-(苄氧基)-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(55.0mg,0.087mmol)通过prep-chiral-hplc用下列条件分离:(柱:chiralpak id,2x25cm(5μm);流动相a:甲基叔丁基醚(加上0.2%异丙胺),流动相b:etoh;流率:17ml/min;梯度:10%b,13min;检测器:uv 220/254nm),以得到呈淡黄色固体状的顺式-n-(5-(3-(苄氧基)-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(实施例572,rt1:8.70min)(15.3mg,28%):1h nmr(400mhz,dmso-d6)δ8.90(s,1h),8.29(s,1h),8.09(d,j=8.3hz,1h),8.02(d,j=1.9hz,1h),7.98(d,j=12.1hz,1h),7.42-7.34(m,4h),7.32-7.27(m,1h),4.90-4.82(m,1h),4.53(s,2h),4.47(t,j=5.4hz,2h),3.32(s,3h),3.06(s,3h),3.05-2.88(m,5h),2.68(dd,j=13.9,6.2hz,2h),1.08(d,j=6.2hz,6h);ms:[(m 1)]
=634.50和呈无色固体状的反式-n-(5-(3-(苄氧基)-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(实施例567,rt2:11.2min)(12.6mg,23%):1h nmr(400mhz,dmso-d6)δ8.88(s,1h),8.59(d,j=8.3hz,1h),8.33(s,1h),8.02-7.94(m,2h),7.36-7.21(m,5h),4.58(q,j=6.3hz,1h),4.44(t,j=5.4hz,2h),4.41-4.33(m,1h),3.28(s,3h),3.05(s,3h),2.00-3.94(m,3h),2.92-2.84(m,1h),2.83-2.74(m,1h),2.69(dd,j=13.1,5.8hz,2h),2.59-2.54(m,1h),1.35(d,j=6.4hz,3h),1.05(d,j=6.3hz,6h);ms:[(m 1)]
=634.55。
[0263]
化合物573
[0264][0265]
3-氧代环丁烷-1-甲酸癸酯:在环境温度下向3-甲基环丁烷-1-甲酸(3.00g,26.3mmol)和1-癸醇(4.16g,26.3mmol)于二氯甲烷(90.0ml)中的搅拌溶液中添加1-乙基-3-(3-二甲氨基丙基)碳二亚胺盐酸盐(7.56g,39.4mmol)和4-二甲氨基吡啶(0.32g,2.62mmol)。将所得混合物在25℃下搅拌16小时并通过水(30.0ml)猝灭。将所得混合物用二氯甲烷(3x100ml)萃取。将合并的有机层用盐水(2x50.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~8%乙酸乙酯/石油醚洗
脱。将所需级分收集并在减压下浓缩以得到呈淡黄色油状的标题化合物(3.10g,47%):1hnmr(400mhz,cdcl3)δ4.15(t,j=6.7hz,2h),3.47-3.35(m,2h),3.35-3.15(m,3h),1.69-1.61(m,2h),1.39-1.22(m,14h),0.88(t,j=6.7hz,3h);ms:[(m 1)]
=252.20。
[0266][0267]
3-亚乙基环丁烷-1-甲酸癸酯:在14℃下向乙基三苯基溴化鏻(17.3g,46.5mmol)于二甲亚砜(300ml)中的溶液中逐份添加叔丁醇钾(4.94g,44.1mmol)。在氮气气氛下,在25℃下搅拌所得混合物0.5小时,然后在14℃下经2min逐滴添加3-氧代环丁烷-1-甲酸癸酯(8.00g,31.5mmol)。在25℃下搅拌4小时之后,在0℃下将反应通过饱和氯化铵水溶液(20.0ml)猝灭。将所得混合物用水(1.00l)稀释并用乙酸乙酯(3x200ml)萃取。将合并的有机层用盐水(2x100ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用1%~5%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈无色油状的标题化合物(1.70g,21%):1h nmr(400mhz,cdcl3)δ5.22-5.14(m,1h),4.09(t,j=6.7hz,2h),3.10(tt,j=9.2,7.2hz,1h),2.96-2.79(m,4h),1.67-1.58(m,2h),1.49(dq,j=6.0,2.0hz,3h),1.39-1.20(m,14h),0.88(t,j=6.7hz,3h)。
[0268][0269]
3-乙基环丁烷-1-甲酸癸酯:在环境温度下,向3-亚乙基环丁烷-1-甲酸癸酯(700mg,4.94mmol)于甲醇(10.0ml)中的搅拌溶液中添加无水pd/c(70.0mg,10%钯炭)。在环境温度下,在氢气气氛(2atm.)下搅拌16小时之后,将所得混合物过滤。将滤饼用甲醇(3x20.0ml)洗涤。将滤液在减压下浓缩以得到呈淡黄色油状的标题化合物(666mg,95%):1h nmr(400mhz,cdcl3)δ4.10-4.02(m,2h),3.10-2.87(m,1h),2.40-2.22(m,2h),2.16-2.03(m,1h),1.91-1.76(m,2h),1.67-1.56(m,2h),1.48-1.21(m,16h),0.92-0.75(m,6h)。
[0270][0271]
1-(6-溴-7-氟-3-硝基喹啉-4-基)-3-乙基环丁烷-1-甲酸癸酯:在氮气气氛下,在-78℃下向新近制备的二异丙基氨基锂(2.45mmol)于无水四氢呋喃(20.0ml)中的溶液中添加3-乙基环丁烷-1-甲酸癸酯(657mg,2.45mmol)。在-78℃下搅拌所得混合物1小时,然后添加6-溴-4-氯-7-氟-3-硝基喹啉(575mg,1.88mmol)。在25℃下再搅拌1小时之后,将反应通过饱和氯化铵水溶液(10.0ml)猝灭。将所得混合物用水(20.0ml)稀释并分离。将水层用乙酸乙酯(5x30ml)萃取。将合并的有机层用盐水(4x30.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用3%~9%乙酸乙酯/石油醚
洗脱。将所需级分收集并在减压下浓缩以得到呈黄色油状的标题化合物(450mg,粗制的):ms:[(m 1)]
=537.25,539.25。
[0272][0273]
8'-溴-3-乙基-7'-氟螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:在环境温度下搅拌粗制的1-(6-溴-7-氟-3-硝基喹啉-4-基)-3-乙基环丁烷-1-甲酸癸酯(450mg,0.84mmol)和铁粉(327mg,5.86mmol)于乙酸(8.00ml)中的混合物16h。将所得混合物过滤,将滤饼用四氢呋喃(5x300ml)洗涤。在减压下浓缩滤液。将残余物用饱和碳酸氢钠水溶液碱化至ph=8。将所得混合物用乙酸乙酯(5x100ml)萃取。将合并的有机层用盐水(50.0ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过prep-tlc(dcm/meoh=15/1,v/v)纯化以得到呈黄色固体状的标题化合物(40.0mg,14%):1h nmr(400mhz,dmso-d6)δ10.76(d,j=5.3hz,1h),8.67(d,j=1.7hz,1h),8.45(d,j=7.2hz,0.4h),8.39(d,j=7.4hz,0.6h),7.98(dd,j=10.1,3.4hz,1h),2.88-2.73(m,2h),2.55-2.51(m,2h),2.29-2.19(m,1h),1.78-1.64(m,2h),0.99-0.88(m,3h);ms:[(m 1)]
=349.05,351.05。
[0274][0275]
8'-溴-3-乙基-7'-氟-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮:在氮气气氛下,在0℃下向8'-溴-3-乙基-7'-氟螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮(35.0mg,0.10mmol)于n,n-二甲基甲酰胺(3.00ml)中的溶液中添加氢化钠(5.21mg,0.13mmol,60%分散于矿物油中)。将所得混合物在25℃下搅拌30分钟,然后在0℃下添加碘甲烷(21.4mg,0.15mmol)。在25℃下再搅拌40分钟之后,将反应通过饱和氯化铵水溶液(20.0ml)猝灭。将所得混合物由水(100ml)稀释并用乙酸乙酯(4x100ml)萃取。将合并的有机层用盐水(4x100ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过prep-tlc(dcm/meoh=20/1,v/v)纯化以得到呈黄色固体状的标题化合物(30.0mg,83%):1h nmr(400mhz,dmso-d6)δ8.90(d,j=2.9hz,1h),8.49(d,j=7.4hz,0.4h),8.42(d,j=7.4hz,0.6h),8.02(dd,j=10.1,3.1hz,1h),3.29(d,j=4.5hz,3h),2.93-2.75(m,2h),2.61-2.52(m,2h),2.29-2.21(m,1h),1.82-1.67(m,2h),0.98-0.89(m,3h);ms:[(m 1)]
=363.05,365.05。
[0276][0277]
n-(5-(3-乙基-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺:向8'-溴-3-乙基-7'-氟-3'-甲基螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-2'(3'h)-酮(50.0mg,0.14mmol)和n-[2-[2-(异丙氨基)乙氧基]-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-3-基]甲磺酰胺(165mg,0.41mmol)于水(0.75ml)和1,4-二噁烷(3.00ml)中的溶液中添加碳酸钠(17.5mg,0.17mmol)和四(三苯基膦)钯(0)(31.8mg,0.028mmol)。在氮气气氛下,在80℃下搅拌2小时之后,在减压下浓缩所得混合物。将残余物通过prep-tlc(dcm/meoh=10/1,v/v)纯化以得到粗产物,将该粗产物通过反相快速色谱用下列条件进一步纯化:柱:spherical c18,20~40μm,120g;流动相a:水(加上10mm nh4hco3),流动相b:乙腈;流率:45ml/min;梯度(b%):5%~25%,7min;25%~45%,25min;45%~65%,8min;95%,5min;检测器:uv 254nm。将含有所需产物的级分在17min下收集并在减压下浓缩以得到呈黄色固体状的标题化合物(30.0mg,40%):ms:[(m 1)]
=556.20。
[0278][0279]
顺式-n-(5-(3-乙基-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(实施例573)和反式-n-(5-(3-乙基-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(实施例566):将上述n-(5-(3-乙基-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(30.0mg)通过prep-chiral-hplc用下列条件分离:柱:chiralpak ic,2x25cm,5μm;流动相a:己烷/dcm=3/1(加上0.2%异丙胺),流动相b:etoh;流率:20ml/min;梯度:30%b,15min;检测器:uv 220/254nm。将所需级分收集并在减压下浓缩以得到呈无色固体状的顺式-n-(5-(3-乙基-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰胺(实施例573,rt1:10.94min)(12.8mg,43%):1hnmr(400mhz,dmso-d6)δ8.87(s,1h),8.32-8.22(m,2h),7.98(d,j=12.2hz,2h),4.44(t,j=5.3hz,2h),3.30(s,3h),3.04(s,3h),2.99(t,j=5.3hz,2h),2.94-2.78(m,4h),2.26(dd,j=12.2,5.7hz,2h),1.75(t,j=7.2hz,2h),1.07(d,j=6.3hz,6h),0.89(t,j=7.3hz,3h);ms:[(m 1)] =556.25
[0280]
和呈无色固体状的反式-n-(5-(3-乙基-7'-氟-3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)-2-(2-(异丙氨基)乙氧基)吡啶-3-基)甲磺酰
胺(实施例566,rt2:13.63min)(8.6mg,29%):1hnmr(400mhz,dmso-d6)δ8.88(s,1h),8.34-8.26(m,2h),8.01-7.93(m,2h),4.44(t,j=5.4hz,2h),3.31(s,3h),3.05(s,3h),3.00(d,j=5.4hz,2h),2.96-2.83(m,2h),2.70-2.60(m,2h),1.66(q,j=7.2,6.7hz,2h),2.56-2.52(m,2h),1.07(d,j=6.2hz,6h),0.89(t,j=7.3hz,3h);ms:[(m 1)]
=556.20。
[0281]
化合物574
[0282][0283]
1-(5-溴-3-硝基吡啶-2-基)-n,n-二甲基氮杂环丁烷-3-胺:在环境温度下,向n,n-二甲基氮杂环丁烷-3-胺盐酸盐(0.27g,2.08mmol)和5-溴-2-氯-3-硝基吡啶(0.49g,2.08mmol)于四氢呋喃(40.0ml)中的搅拌溶液中添加二异丙基乙胺(0.67g,5.19mmol)。将所得混合物搅拌3小时并在减压下浓缩。将残余物通过硅胶柱色谱纯化,用3%~9%乙酸乙酯/石油醚洗脱。将所需级分收集并在减压下浓缩以得到呈黄色固体状的标题化合物(0.50g,59%):1h nmr(400mhz,cdcl3)δ8.37(d,j=2.2hz,1h),8.29(d,j=2.2hz,1h),4.20(ddd,j=10.0,7.0,1.2hz,2h),3.93(ddd,j=9.9,5.1,1.2hz,2h),3.16(tt,j=7.0,5.1hz,1h),2.20(s,6h);ms:[(m 1)]
=301.00,303.00。
[0284][0285]
1-(5-溴-3-硝基吡啶-2-基)-n,n-二甲基氮杂环丁烷-3-胺。在环境温度下,向1-(5-溴-3-硝基吡啶-2-基)-n,n-二甲基哌啶-4-胺(6.20g,20.7mmol)于乙酸(90.0ml)中的溶液中添加铁粉(11.5g,206mmol)。在环境温度下搅拌所得混合物2小时。将所得混合物过滤,并将滤饼用四氢呋喃(3x100ml)洗涤。在减压下浓缩滤液。将残余物用饱和碳酸钠水溶液(100ml)溶解并用乙酸乙酯(3x100ml)萃取。将合并的有机层用盐水(3x100ml)洗涤并经无水硫酸钠干燥。过滤之后,在减压下浓缩滤液。将残余物通过硅胶柱色谱纯化,用2%~4%甲醇/二氯甲烷洗脱,得到呈灰色固体状的标题化合物(5.20g,92%):1h nmr(400mhz,cdcl3)δ7.74(d,j=2.0hz,1h),6.93(d,j=2.0hz,1h),4.19-4.04(m,2h),3.93-3.88(m,2h),3.21-3.14(m,1h),2.24(s,6h);ms:[(m 1)]
=271.00,273.00。
[0286][0287]
n-(5-溴-2-(3-(二甲氨基)氮杂环丁烷-1-基)吡啶-3-基)甲磺酰胺:在环境温度下,向5-溴-2-[3-(二甲氨基)氮杂环丁烷-1-基]吡啶-3-胺(2.10g,7.77mmol)和n,n-4-二甲氨基吡啶(77.0mg,0.63mmol)于吡啶(70.0ml)中的搅拌溶液中逐滴添加甲磺酰氯(1.77g,15.5mmol)。在环境温度下,在氮气气氛下搅拌3小时之后,在减压下浓缩所得混合
物。将残余物通过反相快速色谱用下列条件纯化:柱:spherical c18,20~40μm,330g;流动相a:水(加上10mm nh4hco3),流动相b:乙腈;流率:65ml/min;梯度(b%):5%~20%,8min;20%~40%,20min;40%~95%,2min;95%,5min;检测器:uv 254nm。将所需级分在19min下收集并在减压下浓缩以得到呈无色固体状的标题化合物(1.40g,52%):1h nmr(400mhz,cd3od)δ7.92(d,j=2.2hz,1h),7.61(d,j=2.2hz,1h),4.27(dd,j=8.8,7.2hz,2h),3.96(dd,j=9.0,5.6hz,2h),3.24-3.16(m,1h),3.00(s,3h),2.20(s,6h);ms:[(m 1)]
=349.00,351.00。
[0288][0289]
n-(2-(3-(二甲氨基)氮杂环丁烷-1-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-3-基)甲磺酰胺:在环境温度下,向n-[5-溴-2-[3-(二甲氨基)氮杂环丁烷-1-基]吡啶-3-基]甲磺酰胺(1.00g,2.86mmol)和4双(频哪醇合)二硼(2.18g,8.59mmol)于1,4-二噁烷(30.0ml)中的溶液中添加乙酸钾(1.12g,11.5mmol)和双(二苯基膦)二茂铁]二氯钯(ii)二氯甲烷加合物(351mg,0.43mmol)。在氮气气氛下,在85℃下搅拌所得混合物2小时。在减压下浓缩所得混合物。将残余物通过反相快速色谱在下列条件下纯化:柱:spherical c18,20~40μm,330g;流动相a:水(加上10mm nh4hco3),流动相b:乙腈;流率:65ml/min;梯度(b%):5%~20%,7min;20%~40%,12min;40%~95%;2min;95%,5min;检测器:uv 254nm。将所需级分在20min下收集并在减压下浓缩以得到呈黄色固体状的标题化合物(800mg,71%):1h nmr(400mhz,dmso-d6)δ8.78(s,1h),8.18(d,j=1.5hz,1h),7.50(d,j=1.6hz,1h),4.19(t,j=8.0hz,2h),3.93(dd,j=8.9,5.0hz,2h),3.12-3.04(m,1h),2.99(s,3h),2.09(s,6h),1.27(s,12h);ms:[(m 1)]
=397.20。
[0290][0291]
n-(2-(3-(二甲氨基)氮杂环丁烷-1-基)-5-(3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)吡啶-3-基)甲磺酰胺:向8-溴-3-甲基-2,3-二氢螺[环丁烷-1,1-吡咯并[2,3-c]喹啉]-2-酮(320mg,1.01mmol)和n-[2-[3-(二甲氨基)氮杂环丁烷-1-基]-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-3-基]甲磺酰胺(600mg,1.51mmol)于1,4-二噁烷(13.0ml)和水(2.00ml)中的溶液中添加碳酸钠(160mg,1.51mmol)和四(三苯基膦)钯(0)(175mg,0.15mmol)。在氮气气氛下,在85℃下搅拌2小时之后,在减压下浓缩所得混合物。将残余物通过prep-tlc(dcm/meoh=8/1,v/v)纯化以得到呈淡黄色固体状的标题化合物:1h nmr(400mhz,dmso-d6)δ9.07(s,1h),8.80(s,1h),8.58(s,
1h),8.33(s,1h),8.13(d,j=8.9hz,1h),7.95(d,j=9.2hz,1h),7.91(d,j=2.1hz,1h),4.28-4.21(m,2h),4.02-3.95(m,2h),3.30(s,3h),3.14(s,4h),2.98-2.90(m,2h),2.60-2.52(m,4h),2.13(s,6h);ms:[(m 1)]
=507.20。
[0292][0293]
n-(2-(3-(二甲氨基)氮杂环丁烷-1-基)-5-(3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)吡啶-3-基)甲磺酰胺盐酸盐:将n-(2-(3-(二甲氨基)氮杂环丁烷-1-基)-5-(3'-甲基-2'-氧代-2',3'-二氢螺[环丁烷-1,1'-吡咯并[2,3-c]喹啉]-8'-基)吡啶-3-基)甲磺酰胺(350mg,0.69mmol)于稀盐酸水溶液(115ml,0.69mmol,0.006m)和乙腈(20.0ml)中的溶液直接冻干以得到呈橙色固体状的标题化合物(375mg,100%):1h nmr(400mhz,dmso-d6)δ10.77(s,1h),9.24(s,1h),8.83(s,1h),8.64(d,j=2.1hz,1h),8.35(s,1h),8.17(d,j=8.9hz,1h),8.02-7.94(m,2h),4.50-4.41(m,2h),4.39-4.32(m,2h),4.27-4.16(m,1h),3.31(s,3h),3.18(s,3h),2.99-2.88(m,2h),2.82(d,j=4.1hz,6h),2.63-2.52(m,4h);ms:[(m 1)]
=507.20。
[0294]
靶向和脱靶抑制活性
[0295]
细胞西方测定中的atm:
[0296]
在早上,将mcf-7乳腺癌细胞以10,000个细胞/孔的密度置于384孔板(corning,#356663)内,每孔25μl细胞。次日,使用pin工具(echo 550)将化合物添加到该板中,经由3倍连续稀释(总共10个剂量)至1μm的最终浓度。然后,添加依托泊苷(sigma,#e1383)至100μm的最终浓度。将板在37℃下孵育1h,并且在环境温度下,通过添加25μl固定溶液(8%多聚甲醛)固定细胞20分钟。用含有0.1%triton x-100的1x pbs(磷酸盐缓冲盐水)洗涤5次,使细胞透化;每次洗涤为5分钟长。通过在环境温度下,于384孔板中在振荡下添加50μl odyssey阻断缓冲液(li-cor,#927-40000)来阻断细胞1.5小时。然后去除阻断缓冲液,并将20μl抗pkap1抗体(bethyl laboratories,#a300-767a)(1/2000)溶液添加到384孔板的每个孔中。将板在4℃下孵育过夜,然后用1x pbst(含有0.1%tween-20的1x pbs)洗涤5次。将含有dna染色draq5(cst,#4084l)(1/5,000)(20μl)的二抗(irdye 800cw山羊抗兔igg,li-cor,#926-32211)(1/5,000)溶液添加到该板的每个孔中,并且在黑暗中在温和振荡下孵育该板1小时。在黑暗中,在环境温度下在温和振荡下用1x pbst(含有0.1%tween-20的1x pbs)洗涤细胞5次。最后一次洗涤之后,去除洗涤溶液,将板倒置于薄的纸巾上并在1000rpm下离心1min以吸收所有的洗涤缓冲液。用潮湿的无绒纸清洁板的底部。立即使用odyssey clx(li-cor)扫描板。
[0297]
dna-pk酶联免疫吸附测定:
[0298]
第一天,在每个孔中用0.1m na2co3/nahco3(ph 9.6)稀释3μggst-p53,以使得用gst-p53(1-101)肽(由pharmaron,bcs department纯化)包被96孔板(thermofisher,目录号:442404)。在4℃下孵育该板过夜。第二天,去除包被缓冲液,并用pbst(含有0.1%tween-20的1x pbs)洗涤板两次。然后添加dna-pk酶溶液(invitrogen,#pr9107a;最终dna-pk浓
度:0.1μg/ml)。将化合物连续稀释至最终最大浓度100nm(3倍系列稀释,总共10个剂量),并将atp溶液(最终atp浓度:20μm)添加到板中。在25℃下孵育该板1小时。将板用pbst(含有0.1%tween-20的1x pbs)洗涤三次并在4℃下用pbst和1%bsa的溶液阻断过夜。第三天,用pbst(含有0.1%tween-20的1x pbs)洗涤该板4次。将抗磷酸p53一抗(cell signaling technology,#9286,phospho-p53(ser15)(16g8)小鼠mab)(1/1000)添加到每个孔中。将板密封,在37℃下孵育1h,并用pbst(含有0.1%tween-20的1x pbs)洗涤4次。将hrp连接的二抗(cell signaling technology,#7076,抗小鼠igg,hrp连接的抗体)(1/1000)(100μl)添加到每个孔中。将板用胶带密封,在37℃下孵育30min,并用pbst(含有0.1%tween-20的1x pbs)洗涤4次。此时,将100μl tmb(cell signaling technology,#7004)底物添加到每个孔中。将板用胶带密封并在37℃下孵育该板10min。将终止溶液(cell signaling technology,#7002)(100μl)添加到每个孔中,并将板在450nm下进行吸收检测。
[0299]
mtor生化测定:
[0300]
mtor激酶反应在低体积384孔板中以10μl的体积进行。通常,使用perkinelmer型号6008260板。1x激酶反应缓冲液的组成是:50mm hepes ph 7.5、0.01%tween 20、1mm egta、10mm mncl2和2mm dtt。添加mtor酶的溶液(thermofisher,#pr8683b;最终mtor浓度:0.5μg/ml),并且将化合物连续稀释至100nm的最终最大浓度(3倍系列稀释,总共10个剂量)。将gfp-4e-bp1(最终浓度:0.4μm)和atp溶液(最终atp浓度:3μm)添加到384孔板中。将板在25℃下孵育1小时,并将10μl于tr-fret稀释缓冲液中的edta溶液(20mm)和tb标记的抗p4e-bp1抗体(4nm)添加到每个孔中。将板密封,在25℃下孵育30min,并在为lanthascreen
tm
trfret配置的读板器上读取。
[0301]
pi3kα和pi3kδ生化测定:
[0302]
将和激酶反应在低体积的384孔板中以5μl体积进行。通常,使用perkinelmer型号6008280板。1x激酶反应缓冲液由以下组成:50mm hepes ph 7.5、3mm mgcl2、0.03%chaps、1mm egta、100mm nacl和2mm dtt。将(thermofisher,#pv4788;最终浓度:120ng/ml)或酶溶液(thermofisher,#pv6451;最终浓度:250ng/ml)添加到板中,将化合物连续稀释至100nm的最终最大浓度(3倍系列稀释,总共10个剂量),并将pip2:3ps(最终浓度:)和atp溶液(最终atp浓度:10μm)添加到384孔板中。在25℃下孵育该板1小时。将adp-glo试剂缓冲液(5μl)添加到每个孔中。将板密封并在25℃下孵育40min。将adp-glo检测缓冲液(10μl)添加到每个孔中,并且将板在25℃下孵育40min并在为luminescence配置的读板器上读取。
[0303]
体外herg抑制测定:
[0304]
通过将tet调控的表达载体pt-rex-dest30中的herg编码序列转染到表达tet阻遏物的细胞(t-rex tm hek293)中由此产生可诱导表达高水平的herg通道的细胞来生成herg-t-rex tm hek 293细胞(invitrogen,k1236)。将细胞在含有85%dmem、10%透析fbs、0.1mm neaa、25mm hepes、100u/ml青霉素-链霉素、5μg/ml杀稻瘟菌素和400μg/ml遗传霉素的培养基中培养。每周使用tryple
tm
express(gibco,12604)分裂细胞约3次,并保持在约40%至约80%的汇合度之间。在测定之前,用1μg/ml的强力霉素(sigma,d9891)诱导细胞48小时。在实验当天,使用前将诱导的细胞重悬并以5
×
105个细胞/6cm细胞培养皿铺于盖玻片上。通
过配备放大器(heka,epc10 and molecular devices,multiclamp 700b)和倒置相差显微镜(olympus,ix51/71/73)的手动膜片钳记录系统获取herg通道介导的电流。玻璃移液管由微量移液管拉拔器(sutter,p97 and narishige,pc-10)制备,并通过在范围内的移液管阻力而合格。内部吸移管溶液是140mm kcl、2mm mgcl2、10mm egta、5mm mgatp和10mm hepes(用koh将ph调节至7.35),且外部缓冲液是132mm nacl、4mm kcl、3mm cacl2、0.5mm mgcl2、11.1mm葡萄糖和10mm hepes(用naoh将ph调节至7.35)。保持全细胞配置,持续监测接入阻力通过将膜去极化至 30mv持续4.8秒来引发herg电流,并将电压设置回-50mv持续5.2秒来去除失活并测量去活化尾电流。尾电流大小的最大量用于测定herg电流幅度。为了评价herg抑制作用,在全细胞记录配置下,通过液体灌注系统(ala,vm8重力流输送系统)将空白媒介物和测试物品灌注到细胞中。对于剂量响应测定,将测试物品从低浓度至高浓度累积地施加于细胞。实验中使用了阳性对照(多非利特),以确保作为方法验证的主要部分的细胞和操作的性能。针对剂量浓度拟合herg电流抑制百分比以建立剂量响应曲线并确定ic
50
。
[0305]
在一些实施方案中,本发明化合物选自由下表中列出的化合物组成的组。
[0306]
表1测定结果
[0307]
[0308][0309]
克隆源性和体内功效测定
[0310]
方法
[0311]
细胞培养.mcf-7人乳腺癌细胞系及hct116 p53野生型和hct116 p53-/-细胞系是从atcc处获得。将mcf7和hct116细胞在杜尔贝科氏改良的伊戈尔培养基(dmem(gibco#11995-065)中培养。mda-mb-231人三阴性乳腺癌细胞系和fadu人头颈鳞状细胞癌细胞系是从charles river laboratories(morrisville,nc)处获得。将mda-mb-231细胞在rpmi 1640培养基(gibco,11875-093)中培养。将fadu细胞在补充有1mm丙酮酸钠(gibco,11360-070)、1x mem非必需氨基酸(gibco,11140-050)的最小必需培养基α(memα)(gibco,12571-063)中培养。所有细胞系均补充有10%胎牛血清(fbs)(corning,35-010-cv)和1x抗生素-抗真菌剂(gibco#15240-062)并在37℃下在5%co2中生长。所有细胞系均通过短串联重复图谱分析验证,并且支原体检测呈阴性。
[0312]
抗体.识别磷酸化/活化atm(s1981,#ab81292)和dna-pkcs(s2056,#ab18192)的抗体购自abcam biotechnology(cambridge,ma)。针对atm(#a1106)和dna-pkcs(#ab1832)的抗体分别来自sigma和abcam。磷酸-kap1(s824,#a300-767a)和kap1(a700-014)抗体来自bethyl labs(montgomery,tx)。磷酸-tbk1(#5483)、cgas(#15102)和psting(s366,#19781s)抗体来自cell signaling technology(cst)(danvers,ma)。sting(pa5-23381)抗体来自thermofisher scientific(waltham,ma),且肌动蛋白(#a5316)抗体来自sigma。所有抗体均以1:1000稀释,例外的是肌动蛋白,其在1xtbst(1x tbs-10x tbs,(corning,46-012-cm)、0.05%tween-20(p7949)sigma和1%bsa中以1:3000稀释。所用的二抗是(a)li-cor方法-山羊抗兔(926-3221)和山羊抗小鼠(926-68020)li-cor;(b)ecl方法:抗兔igg,hrp-连接的(#7074s),cst。
[0313]
免疫印迹.将细胞用20、100、500或1000nm的化合物569或dmso预处理30min,然后用0或10gy(xrad 160,precision x ray)辐照,如图1所示。在图3中,将细胞用以下各物预处理30min:1μm化合物569、1μm atmi(azd0156)、1μm dna-pki(peposertib)作为单一试剂或dmso或与atmi和dna-pki以组合形式(各0.5微摩尔或各1微摩尔),然后用0或10gy(xrad 160,precision x ray)辐照,如图1所示。ir之后1小时,在4℃下,在蛋白酶和磷酸酶抑制剂混合物(roche,#04693159001&#04906837001)存在下将细胞在ripa缓冲液(50nm tris,ph 8.0、150mm nacl、0.1%sds、0.5%脱氧胆酸钠、1%ca-630(sigma,i8896)中裂解20min。将细胞裂解物在4℃下以13,200g离心10min并且使用bca测定(pierce
tm
bca蛋白质测定试剂盒,目录号23227,thermofisher waltham,ma02451)分析上清液的蛋白质含量并且将等量的蛋白质/泳道用于后续的免疫印迹分析。将蛋白质通过添加含有2.5%的最终浓度的β-巯基乙醇的nupage lsd样品缓冲液[4x](invitrogen,np0007)来变性,并使样品沸腾5分钟。将样品装载到nupage 3-8%tris-乙酸蛋白凝胶(invitrogen,ea03785box)中并用1x tris-乙酸sds运行缓冲液(invitrogen,la0041)进行电泳,或者装载到nupage 4-12%bis-tris蛋白凝胶(invitrogen,np0336box)中并用1x mops sds运行缓冲液(invitrogen,np0001)进行电泳。
[0314]
将蛋白质过夜转移到硝酸纤维素膜(amersham,10600003)。将膜用5%脱脂乳粉在1x tbst中阻断1小时并在4℃下用一抗孵育过夜。在rt下将膜用1x tbst洗涤4x10min,然后在rt下用二抗孵育1小时,继之在rt下用1x tbst洗涤3x10min。使用odyssey clx系统或ecl(ecl印迹底物,pierce,32209,34075,34095)对膜进行可视化。
[0315]
克隆源性生存测定.用化合物569进行此测定。将细胞以不同的密度铺于6孔板中:
对于无ir对照,250个细胞;对于2gy的ir,5000个细胞;及对于4gy的ir,10000个细胞,并且培养过夜。第二天,将细胞用100、250、500或1000nm的化合物569或dmso预孵育30分钟,然后暴露于增加剂量的ir(0、2或4gy)。ir之后,将细胞用dmso或化合物569连续孵育5h,之后移除培养基并将细胞用pbs洗涤。将细胞在不存在抑制剂的情况下在完全培养基中培养9天。然后将细胞染色(pbs,0.0037%v/v甲醛,0,1%结晶紫),用水冲洗并干燥。
[0316]
肿瘤研究.雌性ncr nu/nu小鼠(crl:nu(ncr)-foxn1nu,品系490)在5-6周龄时获自charles river。这些研究中使用的所有动物程序均由杜克大学(duke university)的机构动物护理和使用委员会(iacuc)批准。
[0317]
在对数期生长期间收获fadu或mda-mb-231细胞用于体内植入。在制备pbs中的工作稀释液之前,将重悬的细胞在磷酸盐缓冲盐水(pbs)中洗涤3次。将fadu细胞稀释至1x107个细胞/ml,并将mda-mb-231细胞稀释至5x107个细胞/ml。用100μl的细胞悬液在每只小鼠的右侧皮下注射。监测肿瘤生长,直到肿瘤达到至少100mm3的目标体积,此时将小鼠随机分为四个治疗组。
[0318]
在植入肿瘤细胞系后,每周监测一次小鼠的肿瘤进展。检测后,使用数显卡尺二维测量肿瘤。肿瘤在达到至少100mm3后进行治疗,并在招募到治疗组的一个中后每周测量2至3次。研究终点被定义为治疗时肿瘤体积增加了五倍。
[0319]
此研究中使用的媒介物是溶于去离子水中的0.5%(羟丙基)甲基纤维素(hpmc)和0.2%tween80。将媒介物的ph调节至7.0-8.0并将媒介物储存在4℃下。在体内治疗的每一天,在室温下将适量的测试化合物添加到媒介物中。如果必需,将混合物涡旋以产生1.6mg/ml的最终浓度。
[0320]
药效学(pd)测定.如下进行肿瘤研究。向携带fadu或mda-231肿瘤的小鼠预给予单独的媒介物或单独的化合物569(3、6和10mg/kg)。然后在施用媒介物或化合物569后45分钟用10gy辐照肿瘤或不进行辐照(媒介物和化合物569,10mg/kg)。放射后1h采集肿瘤。
[0321]
如下收集和处理用于药效学分析的组织匀浆。将来自fadu或mda-mb-231肿瘤的肿瘤组织在液氮中快速冷冻,并使用组织粉碎机在干冰上粉碎。将一部分肿瘤粉末转移到微管中,然后添加500μlripa缓冲液[50nm tris,ph 8.0、150mm nacl、0.1%sds、0.5%脱氧胆酸钠、1%ca-630(sigma,i8896)]。向10ml的ripa缓冲液中添加2个片剂,每个含有蛋白酶抑制剂和磷酸酶抑制剂(roche,#04693159001&#04906837001)、200μl 0.1m pmsf和160μl抑肽酶(sigma,a6279)。将混悬液在冰上用团粒杵电动机(kontes)均质化并在冰上孵育30分钟。将细胞裂解物在4℃下以13,200rpm离心15min并且将上清液转移至新管中并用bca试剂盒(pierce,23227)分析蛋白质浓度,然后进行免疫印迹。
[0322]
如下进行体内肿瘤生长延迟研究。在植入肿瘤细胞系后,每周监测一次小鼠的肿瘤进展。在测量到至少100mm3的肿瘤进展之后,将携带肿瘤的小鼠分到4个组的一个中,每组使用5只小鼠。肿瘤体积介于100与500mm3之间。媒介物或化合物569(10mg/kg)每天一次通过口服管饲法施用,连续三天(qd
×
3),单独或与局灶性放射组合。基于各个动物的体重调整媒介物或化合物569的递送体积。分到放射疗法组的小鼠在施用媒介物或化合物569之后45分钟每天接受3gy,连续三天(qd
×
3)。在放射治疗期间用异氟烷麻醉小鼠,放射治疗是使用x-rad 225cx小动物图像引导辐照器(precision x-ray)进行的。通过荧光检查,40mm x 40mm的辐照场以携带肿瘤的腿为中心。使用0.3mm cu过滤器以300cgy/min的平均剂量率
对小鼠进行225kvp、13ma x射线的平行相对前场和后场辐照。
[0323]
第1组小鼠接受媒介物,qd
×
3。
[0324]
第2组小鼠接受10mg/kg化合物569,qd
×
3。
[0325]
第3组小鼠在3gy局灶性辐照前45分钟接受媒介物,qd
×
3。
[0326]
第4组小鼠在3gy局灶性辐照前45分钟接受10mg/kg化合物569,qd
×
3。
[0327]
体内数据的统计和图形分析.prism(graphpad)用于图形表示。sas 9.4和r 3.6.1用于统计分析。比较4个治疗组中的肿瘤生长速率。为了在治疗开始时调整不同的肿瘤大小,通过将每个时间点的肿瘤体积除以基线体积来计算相对肿瘤体积。线图用于展示随时间变化的中位相对肿瘤体积(图6和8)。当动物因肿瘤五倍而退出研究时,为该动物记录的最终肿瘤体积包含在用于计算随后时间点的中位体积的数据中。卡普兰-梅尔绘图用于显示每个治疗组中随时间流逝而留在研究中的小鼠的百分比(图7和9)。在达到五倍终点之前被发现死亡的小鼠受到右删失。用对数秩检验评估四个治疗组之间无五倍生存的差异。p值小于0.05被认为是统计上显著的。
[0328]
结果
[0329]
图1示出了来自评估化合物569在有或没有放射下对mcf7细胞的作用的体外实验的免疫印迹的图像。免疫印迹示出了化合物569在肿瘤细胞中对atm和dna-pk激酶的放射诱导的自磷酸化和kap1(atm底物)的放射诱导的磷酸化的抑制。将mcf7(一种人乳腺癌细胞系)用浓度为20、100、500和1000nm的化合物569预处理30分钟。dmso用作阴性对照。然后将细胞暴露于0或10gy的电离辐射(ir),并且在一小时后,收获细胞用于免疫印迹分析。化合物569对ser2056处dna-pkcs和ser1981处atm的放射诱导的磷酸化(两者皆为自磷酸化事件)和atm底物kap1(ser824)的放射诱导的磷酸化显示出有效的剂量依赖性抑制,证明化合物569在细胞中抑制dna-pk和atm激酶两者。
[0330]
图2展示了化合物569在体外克隆源性生存测定中的放射增敏特性。将mcf7细胞用媒介物或化合物569以100、250、500和1000nm预处理30分钟,然后用增加剂量的ir(0、2和4gy)进行辐照。5h后,移除培养基,并且向细胞中添加不含抑制剂的新鲜培养基,并将其培养9天,然后固定、染色并进行菌落计数。在没有ir的情况下,mcf7细胞中细胞对化合物569的5小时暴露诱导显著程度的放射增敏,而没有细胞毒性的证据(图2,表1)。
[0331]
图3是示出了化合物569、azd0156(选择性atm抑制剂;atmi)、peposertib(选择性dna-pk抑制剂;dna-pki)以及azd0156与peposertib的组合(ai di)在表达野生型p53的hct116细胞或p53表达呈阴性的hct116细胞中诱导tbk1磷酸化的免疫印迹。tbk1的磷酸化是i型干扰素响应激活的标志物,并且与肿瘤对免疫检查点阻断疗法的响应增强有关。通过将两种选择性化学抑制剂(azd0156和peposertib)组合在一起对atm和dna-pkcs的双重抑制导致tbk1的激活比单独使用任何一种选择性抑制剂更深。类似地,将细胞暴露于化合物569(即atm和dna-pkcs的双重抑制剂)是比任何一种单靶向激酶抑制剂更有效的tbk1磷酸化激活剂,并且与p53状态无关。569对tbk1的激活以及atm和dna-pkcs的双重抑制似乎不依赖于cgas和磷酸-sting诱导。
[0332][0333]
为了评估化合物569在体内的药效学活性,fadu和mda-mb-231细胞在裸小鼠中作为肿瘤异种移植物生长。媒介物或化合物569通过口服管饲法单独或与局灶性放射组合施用。向小鼠给予媒介物或化合物569(3、6和10mg/kg)。然后肿瘤在45分钟后接受10gy的局灶性辐照或不进行辐照(0gy,媒介物和0gy 10mg/kg的化合物569)。放射后1h采集肿瘤。来自这些肿瘤的裂解物的免疫印迹显示放射诱导的pdna-pk自磷酸化和放射诱导的kap1(atm的下游靶标)磷酸化以及在放射加化合物569治疗的fadu和mda231肿瘤中对那些磷酸化事件的剂量依赖性抑制。在10mg/kg下,放射诱导的dna-pk自磷酸化和放射诱导的kap1磷酸化的肿瘤水平分别显著抑制了约55%和》80%(图4和5,表2-5)。dna-pk的基础磷酸化在肿瘤中也受到化合物569的抑制。
[0334]
表1在mcf7细胞中的化合物569克隆源性测定(参见图2)
[0335][0336]
表2在fadu肿瘤中pdna-pk/dna-pk的量化
[0337]
治疗pdna-pk/dna-pk标准偏差无放射,媒介物1.140.263无放射,10mg/kg化合物5690.343na10gy,媒介物10.056510gy,3mg/kg化合物5690.9380.18110gy,6mg/kg化合物5690.6580.12610gy,10mg/kg化合物5690.4760.188
[0338]
表3在fadu肿瘤中pkap1/kap1的量化
[0339]
治疗pkap1/kap1标准偏差无放射,媒介物0.005050.000265无放射,10mg/kg化合物5690.00595na10gy,媒介物10.061610gy,3mg/kg化合物5690.09030.00510gy,6mg/kg化合物5690.09820.074110gy,10mg/kg化合物5690.1560.171
[0340]
表4在mda-231肿瘤中pdna-pk/dna-pk的量化
[0341]
治疗pdna-pk/dna-pk标准偏差无放射,媒介物0.6480.0948无放射,10mg/kg化合物5690.223na10gy,媒介物10.23110gy,3mg/kg化合物5690.8890.41410gy,6mg/kg化合物5690.6700.27810gy,10mg/kg化合物5690.4540.116
[0342]
表5在mda-231肿瘤中pkap1/kap1的量化
[0343]
治疗pkap1/kap1标准偏差无放射,媒介物0.003570.000826无放射,10mg/kg化合物569-0.00319na10gy,媒介物10.11410gy,3mg/kg化合物5690.3270.15810gy,6mg/kg化合物5690.01300.0087910gy,10mg/kg化合物5690.009010.00242
[0344]
fadu肿瘤.第1组中的小鼠接受媒介物,qd
×
3。此组中的聚集性肿瘤生长是进行性的。到终点的中位时间为14天,范围为11-14天(图6和7)。第2组中的小鼠接受10mg/kg剂量的化合物569,qd
×
3。到终点的中位时间为14天,范围为11至17天。第2组动物的肿瘤生长曲线和无五倍生存几乎与第1组相同(图6和7)。第3组中的小鼠接受媒介物和3gy的局灶性放射剂量,在施用媒介物后45分钟递送。媒介物和放射皆为qd
×
3。在第16天发现一只动物由于未知原因死亡。到终点的中位时间为21天,范围为19至25天。与第1组和第2组相比,总生存率显著提高(p《0.01,对数秩)。相对于第1组和第2组,在第3组中的聚集性肿瘤生长的延迟和无五倍生存的增加示于图6和7中。第4组中的小鼠接受化合物569和3gy的局灶性放射剂量,在施用化合物后45分钟递送。化合物569和放射皆以qd
×
3施用。到终点的中位时间为42天,范围为31至51天。与第1组、第2组和第3组相比,总生存率显著提高(p《0.01,对数秩)。相对于所有其他组,在第4组中的聚集性肿瘤生长的延迟和无五倍生存的增加示于图6和7中。所有治疗均耐受良好。
[0345]
mda-mb-231肿瘤.第1组中的小鼠接受媒介物,qd
×
3。到终点的中位时间为12天,范围为12至17天。第2组中的小鼠接受10mg/kg剂量的化合物569,qd
×
3。到终点的中位时间为17天,范围为12至22天。第2组动物的肿瘤生长曲线和无五倍生存几乎与第1组相同(图8
和9)。第3组中的小鼠接受媒介物和在施用媒介物后45分钟递送的3gy的局灶性放射剂量。媒介物和放射皆为qd
×
3。到终点的中位时间为22天,范围为16至22天。与用媒介物单独治疗的第1组动物相比,总生存率显著提高(p《0.01,对数秩)。相对于第1组和第2组,在第3组中的无五倍生存的增加示于图9中。第4组中的小鼠接受10mg/kg的化合物569和在施用化合物后45分钟递送的3gy的局灶性放射剂量。化合物569和放射皆为qd
×
3。在第35天发现一只动物由于未知原因死亡。到终点的中位时间为43天,范围为37至58天。与第1组、第2组和第3组相比,总生存率显著提高(p《0.01,对数秩)。相对于所有其他组,在第4组中的聚集性肿瘤生长的延迟和无五倍生存的增加示于图8和9中。所有治疗均耐受良好。
[0346]
其他实施方案
[0347]
在不脱离本发明的范围和精神下,对所述的发明的各种修改和变动将为本领域技术人员显而易见。虽然已联系具体的实施方案描述了本发明,但应理解,所要求保护的发明不应当不适当地限于这些具体的实施方案。实际上,对于本领域技术人员来说显而易见的是,可在本发明的范围内对本发明所描述的方式进行各种修改。
[0348]
其他实施方案在权利要求中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。