1.本发明属于化学合成制备技术领域,涉及化合物的不对称催化,具体涉及一种左旋烟碱的不对称合成方法,不对称氢化一锅法制备高光学活性的四氢吡咯中间体为该发明中的关键步骤。
背景技术:
2.左旋烟碱广泛存在于烟草植株和各种茄科植物中,是一种含吡啶和四氢吡咯环的手性胺类生物碱,由于其特殊的结构而具有独特的生理活性。一方面在农业生产中,烟碱类化合物是一种广泛使用的杀虫剂;另一方面在医药领域,已有临床研究表明,烟碱能作用于乙酰胆碱受体有望成为治疗老年痴呆症、帕金森症、精神分裂症和抑郁症等其他的中枢神经系统疾病的有效药物。除此之外,在化学合成领域中,有研究报道,左旋烟碱还可以作为一种手性离子液体参与各种不对称化学反应。
3.经科学研究证实,左旋烟碱对乙酰胆碱受体的亲和力是右旋烟碱的10-100倍,在市场上其应用也更为广泛。而目前市场上所用的左旋烟碱主要来源于植物提取,其来源受到了原材料、气候以及周期等诸多方面因素的影响,仅仅依靠从植物中提取左旋烟碱已经不能满足市场的需要。因此,借助化学合成制备技术去实现左旋烟碱的大规模生产具有重要的意义。
[0004][0005]
烟碱的化学合成研究一直是科学家们所关注的焦点。天然烟碱在1828年首次由德国化学家posselt和reimann从烟草中分离出来,并于1904年由a.pictet和crepieux首次在实验室中用合成的方式得到。经过了一百多年的发展,出现了很多化学制备左旋烟碱的研究工作报道。现有的左旋烟碱的化学合成方法主要分为两大类,第一类是先合成消旋烟碱,再通过手性拆分的方法得到左旋烟碱,这种方法合成步骤简单,但是需要使用大量的手性拆分试剂使分离纯化操作变得复杂,而且成本较高。参见例如:文献journalof organic chemistry,1990,55,1736-1744;文献journal of the chemical society,perkin transactions i,2002(2),143-154;文献synlett,2009(15),2497-2499;文献journal of heterocyclic chemistry,2009,46(6),1252-1258;专利cn 102617547a;专利cn 107406411a等。
[0006]
第二类是直接通过不对称合成的方法得到左旋烟碱,不需要额外的手性拆分试剂,可以直接获得光学活性的烟碱,但这些方法用于大规模制备左旋烟碱是非常昂贵的,尚未出现商业化的合成路线。例如:文献journalof organic chemisry,1982,47,1069-1073;chavdarian等首次报道了左旋烟碱的不对称合成工作(反应式1)。他们以l-proline为初始原料制得手性氨基醇的模块,再通过五步反应得到了目标产物(s)-nicotine,然而其ee值
仅有24%。
[0007]
反应式1:
[0008][0009]
文献:organic&biomolecular chemistry,2005,3,3266-3268;helmchen等通过金属铱催化烯丙基还原胺化的策略完成了(s)-nicotine的不对称合成,其ee值高达99%(反应式2)。
[0010]
反应式2:
[0011][0012]
文献:journaloforganic chemistry,2011,76(15),5936-5953;o’brien等通过锂化、转金属、金属钯催化的negishi偶联反应,从简单易得的原料n-boc-四氢吡咯出发,完成了(s)-nicotine的不对称合成,其ee值高达84%(反应式3)。
[0013]
反应式3:
[0014][0015]
专利:cn 104341390a;该工作用一种铱-膦噁唑啉手性催化剂催化含吡啶基团的
环状亚胺,以很高的ee值得到关键手性中间体,再通过两步反应得到(s)-nicotine,其ee值高达98%(反应式4)。
[0016]
反应式4:
[0017][0018]
总之,现有的不对称合成左旋烟碱方法,不但所用试剂价格昂贵,而且需要采用低温反应,反应步骤多,分离纯化操作复杂,增加了生产成本和设备成本,很难用于工业化生产。
技术实现要素:
[0019]
鉴于目前合成左旋烟碱的方法存在诸多不足,本发明公开一种左旋烟碱的不对称合成方法,通过不对称催化氢化反应制备高光学活性的四氢吡咯环,再经甲基化就可以得到目标产物左旋烟碱(nicotine)。它是一条原子经济性高,绿色无污染的合成路线,能够大大降低三废量,利于工业放大生产。
[0020]
本发明提供一种左旋烟碱的不对称合成方法,通过以下技术方案来实现:
[0021]
一种左旋烟碱中间体下式(3)的不对称合成方法,其反应路线为:
[0022][0023]
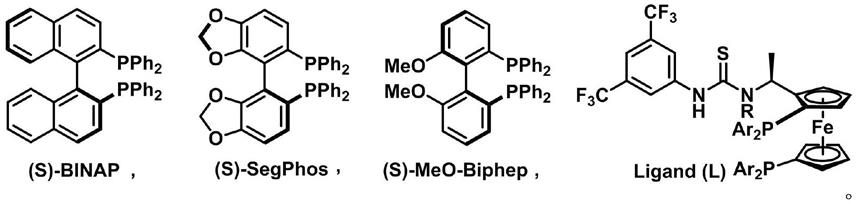
中间体(2)在手性催化剂ir catalyst存在情况下,充入氢气反应得到氢化产物(3),其中,所述ir catalyst由配体和铱金属前体原位络合得到,配体的结构选自:
[0024][0025]
作为本发明的一种优选实施方案,所述ir catalyst由配体和铱金属前体(例如[ir(cod)cl]2)原位络合得到,配体的结构为具有如下结构的二茂铁配体,
所述配体选自具有以下r和ar基团组合的化合物la~lg,
[0026]
la:r=h,ar=ph,zhaophos;
[0027]
lb:r=me,ar=ph;
[0028]
lc:r=h,ar=p-me-ph;
[0029]
ld:r=h,ar=p-meo-ph;
[0030]
le:r=h,ar=3,5-di-me-ph;
[0031]
lf:r=h,ar=3,5-di-me-4-meo-ph;
[0032]
lg:r=h,ar=3,5-di-meo-ph。
[0033]
作为本发明的一种优选实施方案,所述反应在含有二氯甲烷、甲苯、乙酸乙酯的一种或任意比例的混合溶剂中进行。
[0034]
作为本发明的一种优选实施方案,所述反应的温度为20-60摄氏度。
[0035]
作为本发明的一种优选实施方案,所述反应的氢气压力为2-8mpa。
[0036]
作为本发明的一种优选实施方案,所述反应时间为20-60小时。
[0037]
作为本发明的一种优选实施方案,所述中间体(2)与催化剂的摩尔比为2mmol:0.05-1nmol。
[0038]
本发明进一步提供了一种左旋烟碱的不对称合成方法,合成路线如下:
[0039]
其中,所述中间体(3)通过前述的合成方法制备得到。
[0040]
作为本发明的一种优选实施方案,所述的一种左旋烟碱的不对称合成方法,其特征在于,合成路线如下:
[0041][0042]
包括以下步骤:
[0043]
1)氩气保护下,3-溴吡啶的四氢呋喃溶液在-78℃下与n-buli的正己烷溶液混合,然后与n-boc-2-吡咯烷酮混合反应;室温下用稀盐酸溶液淬灭反应,乙酸乙酯萃取,粗产物干燥,旋干,用硅胶层析柱纯化得到中间体(2);
[0044]
2)将中间体(2)和hcl的乙醚溶液常温搅拌反应,抽干溶剂,随后将反应混合物转移至手套箱中,加入手性催化剂ir catalyst,所述中间体(2)与催化剂的摩尔比为2mmol:0.05-1nmol,加入二氯甲烷、甲苯或乙酸乙酯作为溶剂,用氢气置换反应釜气体三次,最后充入2-8mpa氢气,20-60℃下反应20-60小时,缓慢释放反应釜中的气体,旋干,用硅胶柱层析纯化,得到氢化产物(3),其中ir catalyst选自:
[0045][0046]
更优选配体为具有二茂铁骨架的配体(l),选自化合物la~lg。
[0047]
3)在80℃下,中间体(3)加入甲酸和多聚甲醛溶液反应5小时,冷至室温,加碳酸钾直到反应液呈碱性,乙酸乙酯萃取,减压蒸馏得到(s)-烟碱(4)。
[0048]
本发明相对于现有技术具有以下有益效果包括:
[0049]
(1)本发明成功发展了一种左旋烟碱的不对称合成方法,相比同类制备烟碱的技术更为简洁高效。仅三步反应就能制备左旋烟碱,并且能做到优异的立体控制,ee值高达98%,而且操作简便可靠。
[0050]
(2)通过大量的实验研究发现,使用优选的ir catalyst催化不对称氢化反应,反应有非常优异的反应活性和选择性,不需要额外的添加剂,催化剂转化数(ton,turnovernumber)高达5000。
[0051]
(3)本发明操作稳定,纯度高,成本低,有工业放大的前景。
具体实施方式
[0052]
下面结合具体实施例对本发明做进一步的说明,但本发明不局限于此。
[0053]
实施例中未注明具体条件的实验方法,通常按照常规条件以及手册中所述的条件,或按照制造厂商所建议的条件;所用材料、试剂等,如无特殊说明,均可从商业途径得到。
[0054]
实施例1四氢吡咯中间体化合物(3)的制备
[0055][0056]
向中间体2(0.53g,2mmol)中加入4ml hcl的乙醚溶液(2m)和10ml二氯甲烷溶液,在室温下搅拌6小时,反应结束后减压除去溶剂,将反应混合物转移至手套箱中。
[0057]
在氩气氛围下,向反应混合物中加入已配好的催化剂溶液(2ml,0.002mmol,zhaophos和[ir(cod)cl]2的金属络合物催化剂),再加入8ml二氯甲烷,将反应管置于高压釜中,用氢气置换高压釜中的气体三次,最后充入30atm氢气,在25℃下反应24小时。反应结束后,缓慢释放高压釜中的气体,加入20ml饱和碳酸钠溶液中和反应液,用60ml二氯甲烷分三次萃取反应液,收集有机相,再用无水硫酸钠干燥,减压浓缩后用硅胶柱层析纯化,得到氢化产物3。氢化产物3,淡黄色液体,96%yield,98%ee.
[0058]
[α]
25d
=-36.0(c=2.2,meoh),1h nmr(400mhz,cdcl3)δ8.55(d,j=2.1hz,1h),8.42(dd,j=1.8,5.0hz,1h),7.67(dt,j=1.8,7.9hz,1h),7.20(ddd,j=0.6,4.7,7.9hz,1h),4.11(t,j=7.6hz 1h),3.15(ddd,j=5.6,7.6,10.0hz,1h),3.04-2.95(m,1h),2.22-2.13(m,1h),1.97-1.77(m,2h),1.66-1.57(m,1h).
13
c nmr{1h}(101mhz,cdcl3)δ148.5,148.1,140.0,134.0,123.2,59.9,46.8,34.2,25.4.
[0059]
实施例2四氢吡咯中间体化合物(3)的制备
[0060][0061]
在手套箱中,称取双膦配体(0.011mmol),[ir(cod)cl]2(3.4mg,0.005mmol),加入1ml超干溶剂,室温搅拌40分钟,配制成浓度为0.01mol/l催化剂金属络合物溶液,该催化剂溶液可以直接用于均相催化氢化反应。
[0062]
向中间体2(0.53g,2mmol)中加入4ml hcl的乙醚溶液(2m)和10ml二氯甲烷溶液,在室温下搅拌6小时,反应结束后减压除去溶剂,将反应混合物转移至手套箱中。
[0063]
在氩气氛围下,向上述反应混合物中加入已配好的催化剂溶液(100μl,0.001mmol),再加入0.9ml溶剂,将反应管置于高压釜中,用氢气置换高压釜中的气体三次,最后充入60atm氢气,在一定温度(t℃)的恒温油浴中反应48小时。反应结束后,缓慢释放高压釜中的气体,反应混合液用硅胶短柱纯化,浓缩滤液真空干燥得到油状液体,即化合物3,hplc测得转化率和ee值,结果如下表1所示。
[0064]
表1.
[0065][0066][0067]
实施例3(配体la同类催化剂衍生物考察)
[0068][0069]
在手套箱中,称取双膦配体(0.011mmol),[ir(cod)cl]2(3.4mg,0.005mmol),加入1ml超干溶剂,室温搅拌40分钟,配制成浓度为0.01mol/l催化剂金属络合物溶液,该催化剂溶液可以直接用于均相催化氢化反应。
[0070]
向中间体2(0.53g,2mmol)中加入4ml hcl的乙醚溶液(2m)和10ml二氯甲烷溶液,在室温下搅拌6小时,反应结束后减压除去溶剂,将反应混合物转移至手套箱中。
[0071]
在氩气氛围下,向上述反应混合物中加入已配好的催化剂溶液(100μl,浓度见表2),再加入0.9ml溶剂dcm,将反应管置于高压釜中,用氢气置换高压釜中的气体三次,最后充入60atm氢气,在温度(t℃)25℃的恒温油浴中反应48小时。反应结束后,缓慢释放高压釜中的气体,反应混合液用硅胶短柱纯化,浓缩滤液真空干燥得到油状液体,即化合物3,hplc测得转化率和ee值,结果如下表1所示。
[0072]
表2.
[0073][0074][0075]
实施例4中间体2的合成
[0076][0077]
氩气保护下,向三口圆底烧瓶中加入3-溴吡啶(3.16g,20mmol)并用50ml无水四氢呋喃溶解,低温槽中搅拌冷却至-78℃,然后缓慢滴加4.8ml正丁基锂(2.4m)的正己烷溶液,滴加过程中保持-78℃,滴加完后继续保持-78℃搅拌30min,将n-boc-2-吡咯烷酮(3.70g,20mmol)用30ml四氢呋喃溶解,随后滴加至反应混合液中,继续保持-78℃搅拌3小时,然后缓慢升至室温反应24h,用20ml稀盐酸(2m)淬灭反应,乙酸乙酯萃取,有机相用饱和碳酸氢钠和饱和食盐水洗涤,无水硫酸钠干燥,旋干,得粗产品并用乙醚重结晶得中间体2。中间体2,白色固体,70%yield。
[0078]
实例5左旋烟碱的合成
[0079][0080]
中间体3(1.48g,10mmol)加入18ml 88%甲酸和9.2ml 37%甲醛的混合溶液。混合物在80℃下反应5h,然后冷却至室温,加入固体碳酸钾直至反应液为碱性(ph=10-11),用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥,旋干,减压蒸馏得目标产物左旋烟碱。左旋烟碱,无色油状液体,82%yield,98%ee,[α]
25d
=-98.2(c=1,chcl3),1h nmr(400mhz,cdcl3):δ8.56-8.47(m,2h),7.75-7.67(m,1h),7.27-7.23(m,1h),3.32-3.21(m,1h),3.10(t,j=8.3hz,1h),2.39-2.28(m,1h),2.28-2.19(m,1h),2.17(s,3h),2.04-1.91(m,1h),1.89-1.79(m,1h),1.78-1.66(m,1h).
13
c{1h}nmr(101mhz,cdcl3):δ149.5,148.6,138.6,
134.9,123.6,68.9,57.0,40.3,35.1,22.6.
[0081]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。