1.本发明涉及有机过氧化物的合成领域,具体涉及一种3-(2-羟基-2-丙基)异丙苯过氧化氢及其制备方法。
背景技术:
2.3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp)是一种重要的精细有机中间体,由于在其分子结构中同时含有羟基和氢过氧基团,广泛用于制药、合成树脂等精细化工行业。jps5321125a公开了将m-hhp在硫酸催化下酸解制备间异丙烯基苯酚,us4260831a则公开了间异丙烯基苯酚是合成环氧树脂重要原料。us4568768a公开了,以m-hhp为原料,通过三步反应合成制药、农业化学品和染料行业的重要中间体间羟基苯乙酮。
3.同时,m-hhp不仅可以作为二异丙苯空气(氧气)氧化反应的引发剂,还可以作为合成环氧丙烷或环氧丁烷的环氧化剂。jps63250333a公开了在环烷酸钼催化剂存在下,在甲基异丁基酮溶剂中,通过1-丁烯与hhp的环氧化反应,得到环氧丁烷和1,3-二(2-羟基异丙基)苯(m-dc)。利用氢过氧基及羟基的活性,m-hhp还可以用做合成其它有机过氧化物的原料。
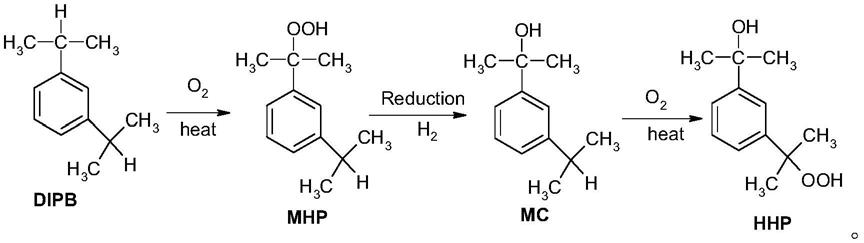
4.传统的m-hhp制备方法,是以纯间位二异丙苯原料,通过空气氧化得到间二异丙苯单氢过氧化物(m-mhp),再通过化学还原剂进行还原或催化加氢得到3-异丙基-二甲基苄醇(m-mc),最后再通过纯氧(或空气)氧化得到m-hhp。其中,生成的含间二异丙苯单氢过氧化物(m-mhp)的氧化液可以通过高真空蒸馏分离得到纯度较高的m-mhp。然而,由于高真空蒸馏的操作对设备要求极高,且过氧化物蒸馏的安全性较差,该方法无法实现大规模工业化生产。
5.虽然,间二异丙苯氧化液还可以采用先与浓氢氧化钠溶液(含量为不低于30重量%)成盐反应,制得m-mhp的钠盐,再通入二氧化碳气体酸化得到纯m-mhp,然后将纯m-mhp还原或催化加氢得到m-mc,再进一步与空气(氧气)进行氧化反应制备得到m-hhp,并且采用碱液萃取、酸化及重结晶得到纯m-hhp。这种合成方法不仅反应步骤多、操作非常复杂,而且过氧化物m-mhp的分离提纯过程存在很大安全隐患,不适合大规模工业生产。
6.gb743210a公开了以189份3-异丙基-二甲基苄醇(m-mc)为原料,在4份氢氧化钙和0.5份偶氮二异丁晴存在下,在90℃油浴加热下,采用纯氧进行氧化反应,当m-mhp含量达到36%时停止反应,冷却反应混合物,加入等体积的苯稀释,过滤除去氢氧化钙,苯溶液用等体积的2n氢氧化钠溶液萃取两次,合并碱液用少量苯洗涤,然后通入二氧化碳进行中和,将析出的白色固体抽滤干燥后得到熔点为71-72℃的m-hhp粗产品。m-hhp粗产品溶解在苯中,过滤除去夹带的碳酸氢钠,在滤液中加入石油醚,析出沉淀,过滤、干燥后得到熔点为71.5-72℃、碘量法测定纯度为99.3%的m-hhp。该方法也存在以下问题:步骤繁多、操作复杂;为了防止mc发生高温脱水副反应,m-mc的分离提纯需要采用高真空蒸馏,对设备要求极高,且过氧化物m-mhp的成盐和分离提纯过程存在很大安全隐患,不适合工业化生产。m-mc的氧气氧化反应转化率不高、产品收率不高,且m-hhp的分离提纯步骤较多,产生大量含碱含盐废
水,对环境保护不利。
7.综上,现有技术的方法存在诸多缺陷,需要一种新的3-(2-羟基-2-丙基)异丙苯过氧化氢的生产方法。
技术实现要素:
8.本发明的目的是为了克服现有技术的3-(2-羟基-2-丙基)异丙苯过氧化氢的生产方法中存在的操作复杂、生产过程不够安全、产品收率较低且纯度较低的问题,提供一种3-(2-羟基-2-丙基)异丙苯过氧化氢及其制备方法,该方法具有产物收率更高且纯度更高的特点。
9.为了实现上述目的,本发明第一方面提供一种3-(2-羟基-2-丙基)异丙苯过氧化氢的制备方法,该方法包括:
10.(1)在溶剂a和极性溶剂b存在下,将1,3-二(2-羟基异丙基)苯和过氧化氢与酸性催化剂接触进行反应;
11.(2)脱除步骤(1)反应得到的产物中的溶剂a;
12.所述溶剂a为烷基芳烃。
13.优选地,1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比为1:1-2,优选为1:1-1.6。
14.本发明提供的方法产物(m-hhp)收率更高,且纯度更高。因此,本发明第二方面提供由第一方面所述的方法制备得到的3-(2-羟基-2-丙基)异丙苯过氧化氢。
15.现有技术中制备3-(2-羟基-2-丙基)异丙苯过氧化氢(本发明又称m-hhp)的方法的反应过程如下:
[0016][0017]
本发明提供的m-hhp制备方法的反应过程如下:
[0018][0019]
相对于现有技术,本发明的方法步骤更简洁,条件温和,易于控制,从而避免了m-mhp及m-mc难以制备和分离的问题,不仅产物收率更高,而且生产过程的安全性更高。同时,本发明的制备过程清洁环保,适合工业化生产。
[0020]
通过上述技术方案,本发明采用特定的1,3-二(2-羟基异丙基)苯(本发明中又称
m-dc),配合特定的溶剂a和溶剂b以及酸性催化剂,与过氧化氢进行反应制备得到m-hhp,制备过程安全性更高,产物收率更高,且纯度更高。
[0021]
在优选情况下,在特定的m-dc与过氧化氢的摩尔比下,配合特定的溶剂a和溶剂b及其特定配比下,使得1,3-二(2-羟基异丙基)苯分子中两个羟基中的一个被氧化,进一步提高了得到的m-hhp产物的收率。
具体实施方式
[0022]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0023]
在本发明中,芳烃是指分子中含有苯环结构的碳氢化合物。
[0024]
在本发明中,所述酸性催化剂是指能够提供质子的化合物。
[0025]
在本发明中,所述真空度表示绝对压强与大气压强的差值。
[0026]
本发明第一方面提供一种3-(2-羟基-2-丙基)异丙苯过氧化氢的制备方法,该方法包括:
[0027]
(1)在溶剂a和极性溶剂b存在下,将1,3-二(2-羟基异丙基)苯和过氧化氢与酸性催化剂接触进行反应;
[0028]
(2)脱除步骤(1)反应得到的产物中的溶剂a;
[0029]
所述溶剂a为烷基芳烃。
[0030]
在本发明中,对过氧化氢的引入方式选择范围较宽,优选地,过氧化氢以双氧水的方式引入,进一步优选地,所述双氧水中过氧化氢的含量为5-30重量%,优选为15-25重量%。在该种优选情况下,更有利于提高产物的收率。
[0031]
根据本发明一种优选实施方式,1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比为1:1-2,优选为1:1-1.6。在该种优选情况下,更有利于提高产物的收率。
[0032]
根据本发明,对所述酸性催化剂的选择范围较宽,只要是能够为所述反应提供质子即可,可以为有机酸,也可以为无机酸。进一步优选地,所述酸性催化剂选自硫酸、高氯酸、硝酸、磷酸和苯磺酸中的至少一种,优选为硫酸和/或高氯酸。在该种情况下,更有利于提高产物的收率。在本发明,所述酸性催化剂可以单独引入,也可以以酸溶液的形式引入,优选以酸溶液的形式引入。本发明对所述酸溶液的浓度选择范围较宽,优选地,所述酸溶液中,酸的含量为15-80重量%,优选为30-60重量%。
[0033]
本发明对所述酸性催化剂的用量选择范围较宽,优选地,所述酸性催化剂与1,3-二(2-羟基异丙基)苯的重量比为0.01-0.1:1;优选为0.01-0.05:1。
[0034]
本发明对所述溶剂a的选择范围较宽,优选地,所述溶剂a的沸点不大于160℃,优选不大于120℃,更优选不大于110℃,例如为40-110℃。在该种优选实施方式下,更有利于提高反应产物的纯度。
[0035]
根据本发明,优选地,所述溶剂a只要是含有烷基芳烃即可,优选地,所述烷基芳烃的碳原子数为7-12,优选为碳原子数为7-9。进一步优选地,所述烷基芳烃为碳原子数为1-6的烃基取代的碳原子数为7-12的芳烃,更优选地,所述烷基芳烃为碳原子数为1-3的烃基取
代的碳原子数为7-9的芳烃。
[0036]
在本发明中,所述碳原子数为1-6的烃基优选为甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基和环己基中的至少一种。在本发明中,所述碳原子数为1-3的烃基优选为甲基、乙基、正丙基和异丙基中的至少一种。
[0037]
根据本发明一种优选实施方式,所述溶剂a选自甲苯、对二甲苯、邻二甲苯、间二甲苯、乙苯和异丙苯中的至少一种。
[0038]
本发明对所述溶剂a的用量选择范围较宽,优选地,相对于10g的1,3-二(2-羟基异丙基)苯,所述溶剂a的用量为100-500ml,优选为150-400ml。
[0039]
在本发明中,溶剂b为极性溶剂,所述极性溶剂的含义为本领域的常规释义。根据本发明一种优选实施方式,所述溶剂b的沸点高于所述溶剂a的沸点,进一步优选地,所述溶剂b的沸点大于150℃,优选大于160℃。
[0040]
根据本发明,优选地,所述溶剂b为碳原子数为2-20的醇、醚和亚砜中的至少一种。
[0041]
根据本发明,优选地,所述溶剂b为碳原子为2-10的醇,优选为碳原子数为2-6的醇。所述醇可以为一元醇,也可以为多元醇,优选为二元醇。在本发明中,所述二元醇是指分子中含有二个羟基的醇类,进一步优选为乙二醇、1,2-丙二醇和1,3-丙二醇中的至少一种。
[0042]
根据本发明,优选地,所述溶剂b为碳原子数为3-18的醚,优选为碳原子数为3-15的醚。优选地,所述醚为二元醇醚,优选为乙二醇醚和/或丙二醇醚。
[0043]
根据本发明,优选地,所述溶剂b为碳原子数为2-8的亚砜,优选为碳原子数为2-4的亚砜。
[0044]
进一步优选地,所述溶剂b选自乙二醇、1,2-丙二醇、1,3-丙二醇、乙二醇醚、丙二醇醚、二甲基亚砜和二乙基亚砜中的至少一种。
[0045]
更优选地,所述溶剂b选自乙二醇、丙二醇醚、二甲基亚砜和二乙基亚砜中的至少一种。在该种优选情况下,更有利于提高反应产物的收率。本发明的发明人发现,通过加入溶剂b,利用所述酸性催化剂与极性溶剂的相互作用,使得反应体系极性增加,所述酸性催化剂能够更好的溶解分散,从而避免了局部酸性过强,保护了生成的氢过氧化物,防止发生产物的酸分解副反应,进而有利于提高产物的收率。
[0046]
进一步更优选地,溶剂b选自乙二醇、乙二醇单甲醚,乙二醇单乙醚,乙二醇单丁醚,乙二醇二乙醚,乙二醇二丁醚,二甘醇,二甘醇单甲基醚,二甘醇单乙醚,二甘醇单丁醚,二甘醇二甲醚,二甘醇二乙醚,二甘醇二丁醚、二甲基亚砜和二乙基亚砜中的至少一种。
[0047]
根据本发明,优选地,所述溶剂b与酸性催化剂的摩尔比为1-25:1,优选为3-10:1。在该种优选情况下,有利于提高产物m-hhp的收率。
[0048]
根据本发明一种优选实施方式,步骤(1)所述反应条件使得以所述反应得到的产物的总量为基准,1,3-二(2-羟基异丙基)苯的含量不大于3重量%,优选为不大于1重量%。在本发明中,反应产物中存在的1,3-二(2-羟基异丙基)苯的含量越少,反应转化率越高。在本发明中,所述反应产物中1,3-二(2-羟基异丙基)苯的含量采用气相色谱测定。
[0049]
根据本发明,优选地,步骤(1)所述反应的条件包括:温度为30-70℃,优选为40-60℃;真空度为-0.095至-0.04mpa,优选为-0.085至-0.07mpa;时间为1-4h,优选为1.5-3h。在该种优选情况下,更有利于提高产物中m-hhp的收率。本发明的发明人发现,所述反应的温度较高时,会导致反应副产物增多,从而降低m-hhp产物的收率;而反应温度较低时,导致反
应速率较慢,降低生产效率。
[0050]
根据本发明,优选地,所述步骤(1)的接触的过程中,还包括引入水(优选为蒸馏水)。在该种优选情况下,可以进一步提高产物的收率。
[0051]
根据本发明,步骤(1)接触过程中,水的引入量选择范围较宽,相对于10g的1,3-二(2-羟基异丙基)苯,水的用量优选为1-100g。
[0052]
根据本发明,优选地,步骤(1)所述反应为回流反应。在该种优选实施方式下,更有利于提高产物的收率。
[0053]
根据本发明一种优选实施方式,所述反应在水和溶剂a共沸状态下进行。在本发明中,优选地,所述溶剂b的沸点高于溶剂a,在该种优选情况下,更有利于在反应过程中除去水。
[0054]
根据本发明一种优选实施方式,将步骤(1)所述反应得到的水和溶剂a的共沸物进行分离,得到的溶剂a回用于步骤(1)进行所述反应。在本发明中,对将共沸物进行分离的设备没有特别的限定,具体地,例如可以在油水分离器中进行。本发明对所述油水分离器没有特别的限定,本领域技术人员可以根据实际需要按需选择。
[0055]
在一种具体地实施方式下,步骤(1)所述反应得到的水和溶剂a的共沸物经冷凝后进入所述油水分离器,水沉入所述油水分离器中进行收集,将油水分离器中得到的溶剂a回用于步骤(1)进行所述反应。
[0056]
根据本发明一种优选实施方式,所述方法包括:在反应的过程中,采用气相色谱测定反应混合物的组成,跟踪控制反应进程,当1,3-二(2-羟基异丙基)苯的含量不大于3重量%时,优选不大于1重量%时,停止所述反应。在该种优选情况下,更有利于提高产物的收率。
[0057]
根据本发明,优选地,步骤(2)采用蒸馏脱除步骤(1)反应得到的产物中的溶剂a。根据本发明,优选地,所述蒸馏之前,步骤(2)还包括:将反应得到的产物进行除水。在本发明中,所述反应得到的产物经冷却后分成水相和油相。优选地,所述步骤(2)除水的过程包括:将步骤(1)中反应得到的产物降温(优选降至室温)后,将分层得到的水相除去。
[0058]
根据本发明,优选地,所述蒸馏之前,所述除水之后,步骤(2)还包括对所述反应得到的产物进行洗涤。在本发明中,对所述洗涤没有特别的限定,所述洗涤剂可以为水。本发明对所述洗涤剂的用量选择范围较宽,以除去所述反应得到的产物中所述酸性催化剂、过氧化氢和所述溶剂b为目的,优选洗涤至洗涤液为ph值为6.5-7。
[0059]
根据本发明,优选地,步骤(2)所述蒸馏的条件包括:温度为30-70℃,优选为40-60℃;真空度为-0.1至0mpa,优选为-0.095至-0.02mpa。在该种优选情况下,减压条件下进行蒸馏,避免了过氧化物的热分解,更有利于提高产物收率和纯度。
[0060]
根据本发明,优选地,该方法还包括步骤(3):将步骤(2)脱溶剂得到的固体产物进行重结晶,得到3-(2-羟基-2-丙基)异丙苯过氧化氢。
[0061]
根据本发明,所述重结晶可以进一步提高产物的纯度。本发明对所述重结晶所采用的结晶溶剂以及结晶条件没有特别的限定,只要有利于提高产物的纯度即可。
[0062]
优选地,所述重结晶采用的结晶溶剂包括溶剂c和溶剂d,所述溶剂c为苯和/或甲苯,所述溶剂d选自正戊烷、正己烷、正庚烷、异辛烷、正葵烷和石油醚中的至少一种。在本发明中,所述结晶溶剂为混合溶剂。在该种优选情况,更有利于提高产物的纯度。
[0063]
在本发明中,反应得到的产物中,m-hhp在所述溶剂c中溶解度大于在溶剂d中的溶解度,优选采用溶剂c和溶剂d的混合结晶溶剂更有利于提高重结晶得到产物的纯度。在本发明中,所述石油醚中包含正戊烷和正己烷,所述石油醚可以通过商购获得。
[0064]
本发明对所述溶剂c和溶剂d的用量选择范围较宽,优选地,所述溶剂c与溶剂d的体积比为1-2:1。在该种优选情况下,更有利于提高产物的纯度。
[0065]
本发明对所述结晶溶剂的用量选择范围较宽,优选地,相对于100重量份的产物3-(2-羟基-2-丙基)异丙苯过氧化氢,结晶溶剂的用量为300-1000重量份,优选为400-600重量份。
[0066]
本发明对所述重结晶的条件选择范围较宽,优选地,所述重结晶的条件包括:温度为0-70℃,优选为10-60℃。
[0067]
根据本发明,优选地,所述重结晶之后,步骤(3)还包括:将重结晶得到的产物进行过滤和干燥。在本发明中,所述过滤可以采用本领域的常规选择,只要能够达到固液分离的目的即可,具体地,例如可以为抽滤或离心分离。
[0068]
根据本发明,优选地,所述过滤之后还包括对过滤得到产物进行洗涤,然后在进行干燥。所述洗涤可以为本领域的常规技术操作,本发明在此不再赘述。
[0069]
根据本发明,优选地,所述干燥的条件包括:温度为40-200℃,优选为50-150℃;时间为0.5-6h,优选为0.5-3h。
[0070]
本发明采用m-dc为原料,在溶剂a和极性溶剂b以及酸性催化剂的同时存在下,通过与过氧化氢的选择性氧化反应,制备得到m-hhp。本发明的方法得到的m-hhp不仅收率更高,且纯度更高。本发明的原料易得,反应条件温和,反应易于控制,反应步骤少,清洁环保,适合大规模工业化生产。
[0071]
因此,本发明第二方面提供由第一方面所述方法制备得到的3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp)。采用本发明的方法制备得到的m-hhp产品收率更高,而且纯度更高。
[0072]
以下将通过实施例对本发明进行详细描述。
[0073]
除非特殊说明,以下实施例中,室温至20℃;
[0074]
以下实施例中,石油醚购买自上海沪试公司,牌号为80098618;
[0075]
以下实施例中,气相色谱的型号为agilent technologies7890b gc system,购买自安捷伦公司。
[0076]
实施例1
[0077]
采用本发明的方法进行3-(2-羟基-2-丙基)异丙苯过氧化氢的制备,步骤如下:
[0078]
(1)在配有搅拌器,温度计,油水分离器和回流冷凝器的三口瓶中,加入200毫升甲苯、10克1,3-二(2-羟基异丙基)苯、9.35克30重量%的双氧水、9.35克去蒸馏水、1克含量为50重量%硫酸及1.58克乙二醇,在50℃水浴加热下,开启搅拌,在真空度约为-0.06mpa下进行反应;
[0079]
其中,1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比为1:1.6;酸性催化剂与1,3-二(2-羟基异丙基)苯的重量比0.05:1;溶剂b与酸性催化剂的摩尔比为5:1;
[0080]
在共沸状态下,水与甲苯经冷却进入油水分离器,其中甲苯返回三口瓶中,而水则沉入油水分离器中收集;反应过程中采用气相色谱检测跟踪反应进程,当测定得到反应混
合物中1,3-二(2-羟基异丙基)苯(m-dc)的含量小于1重量%时,停止反应,反应时间为4小时;
[0081]
(2)将反应产物降温到室温,静置后除去水层,油相用蒸馏水水洗除去硫酸、双氧水及乙二醇,洗涤至洗涤液为中性;然后在50℃下,真空度约为-0.08mpa下,蒸馏除去甲苯,得到脱溶剂产品;
[0082]
(3)将所述脱溶剂产品用苯和正己烷的体积比为1:1的混合溶剂进行重结晶,相对于100重量份的3-(2-羟基-2-丙基)异丙苯过氧化氢,结晶溶剂的用量为500重量份;然后依次抽滤、洗涤和干燥后,得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为78%,测得熔点为71-72℃,结果列于表1。
[0083]
实施例2
[0084]
采用本发明的方法进行3-(2-羟基-2-丙基)异丙苯过氧化氢的制备,步骤如下:
[0085]
(1)在配有搅拌器,温度计,油水分离器和回流冷凝器的三口瓶中,加入150毫升甲苯、10克1,3-二(2-羟基异丙基)苯、7.59克30重量%的双氧水、7.59克去蒸馏水、0.25克含量为50重量%高氯酸及0.97克二甲基亚砜,在60℃水浴加热下,开启搅拌,在真空度约为-0.05mpa下进行反应;
[0086]
其中,1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比为1:1.3;酸性催化剂与1,3-二(2-羟基异丙基)苯的重量比0.0125:1;溶剂b与酸性催化剂的摩尔比为10:1;
[0087]
在共沸状态下,水与甲苯经冷却进入油水分离器,其中甲苯返回三口瓶中,而水则沉入油水分离器中收集;反应过程中采用气相色谱检测跟踪反应进程,当测定得到反应混合物中1,3-二(2-羟基异丙基)苯(m-dc)的含量小于2重量%时,停止反应,反应时间为2.5小时;
[0088]
(2)将反应产物降温到室温,静置后除去水层,油相用蒸馏水水洗后,除去高氯酸、双氧水及二甲基亚砜,洗涤至洗涤液为中性;然后在50℃下,真空度约为-0.08mpa下,蒸馏除去甲苯,得到脱溶剂产品;
[0089]
(3)将所述脱溶剂产品用苯和正己烷的体积比为1:1的混合溶剂进行重结晶,相对于100重量份的3-(2-羟基-2-丙基)异丙苯过氧化氢,结晶溶剂的用量为450重量份;然后依次抽滤、洗涤和干燥后,得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为75%,测得熔点为71-72℃,结果列于表1。
[0090]
实施例3
[0091]
采用本发明的方法进行3-(2-羟基-2-丙基)异丙苯过氧化氢的制备,步骤如下:
[0092]
(1)在配有搅拌器,温度计,油水分离器和回流冷凝器的三口瓶中,加入300毫升甲苯、10克1,3-二(2-羟基异丙基)苯、7.01克30重量%的双氧水、7.01克去蒸馏水、0.5克含量为50重量%硫酸及2.3克乙二醇单乙醚,在40℃水浴加热下,开启搅拌,在真空度约为-0.08mpa下进行反应;
[0093]
其中,1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比为1:1.2;酸性催化剂与1,3-二(2-羟基异丙基)苯的重量比0.025:1;溶剂b与酸性催化剂的摩尔比为10:1;
[0094]
在共沸状态下,水与甲苯经冷却进入油水分离器,其中甲苯返回三口瓶中,而水则沉入油水分离器中收集;反应过程中采用气相色谱检测跟踪反应进程,当测定得到反应混合物中1,3-二(2-羟基异丙基)苯(m-dc)的含量小于1重量%时,停止反应,反应时间为4小
时;
[0095]
(2)将反应产物降温到室温,静置后除去水层,油相用蒸馏水水洗后,除去硫酸、双氧水及乙二醇单乙醚,洗涤至洗涤液为中性;然后在50℃下,真空度约为-0.08mpa下,蒸馏除去甲苯,得到脱溶剂产品;
[0096]
(3)将所述脱溶剂产品用苯和正己烷的体积比为1:1的混合溶剂进行重结晶,相对于100重量份的3-(2-羟基-2-丙基)异丙苯过氧化氢,结晶溶剂的用量为550重量份;然后依次抽滤、洗涤和干燥后,得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为72%,测得熔点为71-72℃,结果列于表1。
[0097]
实施例4
[0098]
采用本发明的方法进行3-(2-羟基-2-丙基)异丙苯过氧化氢的制备,步骤如下:
[0099]
(1)在配有搅拌器,温度计,油水分离器和回流冷凝器的三口瓶中,加入400毫升甲苯、10克1,3-二(2-羟基异丙基)苯、5.84克30重量%的双氧水、5.84克去蒸馏水、0.5克含量为50重量%高氯酸及1.68克二甘醇单乙醚,在55℃水浴加热下,开启搅拌,在真空度约为-0.07mpa下进行反应;
[0100]
其中,1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比为1:1;酸性催化剂与1,3-二(2-羟基异丙基)苯的重量比0.025:1;溶剂b与酸性催化剂的摩尔比为5:1;
[0101]
在共沸状态下,水与甲苯经冷却进入油水分离器,其中甲苯返回三口瓶中,而水则沉入油水分离器中收集;反应过程中采用气相色谱检测跟踪反应进程,当测定得到反应混合物中1,3-二(2-羟基异丙基)苯(m-dc)的含量小于1.5重量%时,停止反应,反应时间为2.5小时;
[0102]
(2)将反应产物降温到室温,静置后除去水层,油相用蒸馏水水洗后,除去高氯酸、双氧水及二甘醇单乙醚,洗涤至洗涤液为中性;然后在50℃下,真空度约为-0.08mpa下,蒸馏除去甲苯,得到脱溶剂产品;
[0103]
(3)将所述脱溶剂产品用苯和石油醚的体积比为1:2的混合溶剂进行重结晶,相对于100重量份的3-(2-羟基-2-丙基)异丙苯过氧化氢,结晶溶剂的用量为600重量份;然后依次抽滤、洗涤和干燥后,得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为70%,测得熔点为71-72℃,结果列于表1。
[0104]
实施例5
[0105]
按照与实施例1相同的方法,不同的是,改变步骤(1)中双氧水的用量,1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比为1:2;
[0106]
得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为65%,测得熔点为71-72℃,结果列于表1。
[0107]
实施例6
[0108]
按照与实施例1相同的方法,不同的是,改变步骤(1)中溶剂b的用量,溶剂b与酸性催化剂的摩尔比为2:1;
[0109]
得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为74%,测得熔点为71-72℃,结果列于表1。
[0110]
实施例7
[0111]
按照与实施例1相同的方法,不同的是,改变步骤(1)中反应温度,在70℃水浴加热
下进行;
[0112]
得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为69%,测得熔点为71-72℃,结果列于表1。
[0113]
实施例8
[0114]
按照与实施例1相同的方法,不同的是,步骤(1)中的酸性催化剂为含量为50重量%的磷酸,用量为1g;
[0115]
其中,酸与1,3-二(2-羟基异丙基)苯的重量比0.05:1;溶剂b与酸性催化剂的摩尔比为5:1;
[0116]
得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为63%,测得熔点为71-72℃,结果列于表1。
[0117]
对比例1
[0118]
按照与实施例1相同的方法,不同的是,步骤(1)中不添加溶剂b;
[0119]
得到3-(2-羟基-2-丙基)异丙苯过氧化氢(m-hhp),收率为37%,测得熔点为65-70℃,结果列于表1。
[0120]
表1
[0121] m-dc:h2o2酸:m-dc溶剂b:酸反应温度/℃m-hhp收率/%实施例11:1.60.05:15:15078实施例21:1.30.0125:110:16075实施例31:1.20.025:110:14072实施例41:10.025:15:15570实施例51:20.05:15:15065实施例61:1.60.05:12:15074实施例71:1.60.05:15:17069实施例81:1.60.05:15:15063对比例11:1.60.05:1/5037
[0122]
注:dc:h2o2表示1,3-二(2-羟基异丙基)苯与过氧化氢的摩尔比;
[0123]
酸:dc表示酸性催化剂与1,3-二(2-羟基异丙基)苯的重量比;
[0124]
溶剂b:酸表示溶剂b与酸性催化剂的摩尔比;
[0125]
m-hhp收率表示产物3-(2-羟基-2-丙基)异丙苯过氧化氢的收率。
[0126]
通过表1的结果可以看出,采用本发明的方法制得的3-(2-羟基-2-丙基)异丙苯过氧化氢收率更高,产物的纯度更高,效果显著。同时,本发明的方法操作简洁,制备过程安全性更高,适合工业化生产。
[0127]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。