1.本发明属于有机合成技术领域,尤其涉及一种盐酸甲氧明的合成方法。
背景技术:
2.盐酸甲氧明为α受体激动剂,有明显的血管收缩作用,它能通过提高外 周阻力,使收缩压和舒张压均升高,而对心脏无兴奋作用;因此,盐酸甲氧 明适用于大出血、创伤、外科手术所引起的低血压及脊髓麻醉前预防低血压 症、室上性阵发性心动过速,也可用于手术后的循环衰竭和因周围循环衰竭 所引起的低血压休克。
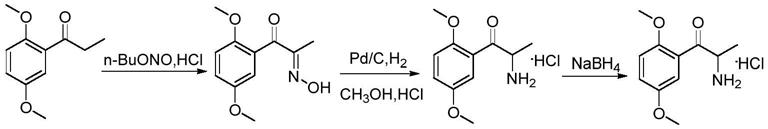
3.现有的盐酸甲氧明工业化合成路线见下式所示,主要以2,5-二甲氧基苯丙酮 为起始物料,与亚硝酸酯类发生肟化反应生成羟肟,再经还原后得到盐酸甲氧 明;该路线起始物料2,5-二甲氧基苯丙酮一般由傅克反应制得,价格较贵,副产 物多难以提纯,肟化反应使用毒性较大的亚硝酸酯类物质,肟化产物稳定性较 差,且反应使用的氯化氢气体对设备腐蚀性较大,中间体使用钯碳还原可能会 引入重金属杂质。因此,开发一条安全高效节能的盐酸甲氧明合成路线很有必 要。
4.
技术实现要素:
5.本发明的目的在于克服上述现有技术的不足之处而提供一种操作简单、反 应条件温和、适用于放大生产的盐酸甲氧明的合成方法。
6.为实现上述目的,本发明采取的技术方案为:一种盐酸甲氧明的合成方法, 所述合成方法包括以下步骤:
7.(1)以2,5-二甲氧基苯甲醛为起始原料,经(a)格式反应后水解,或(b) reformatsky反应后氨解,或(c)reformatsky反应,得3-(2,5-二甲氧基苯基)
ꢀ‑
3-羟基-2-甲基丙酰胺;
8.(2)将3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺经霍夫曼降解反应后 得甲氧明,接着甲氧明成盐,得盐酸甲氧明;
9.本发明的技术方案采用简单易得的2,5-二甲氧基苯甲醛为起始原料,可从 (a)-(c)三条反应路线中任意选择一条合成关键中间体3-(2,5-二甲氧基苯基)
ꢀ‑
3-羟基-2-甲基丙酰胺,再对关键中间体进行霍夫曼降解后成盐即得盐酸甲氧明, 提供的反应要求不苛刻,反应条件温和、反应过程安全可控,能够用于放大生 产。
10.作为本发明所述合成方法的优选实施方式,所述步骤(1)中,(a)格式 反应后水解具体包括以下步骤:将金属催化剂、碘甲烷和卤代试剂加入到有机 溶剂中后于氮气环境下加热回流,接着冷却至室温后加入2,5-二甲氧基苯甲醛反 应,反应结束后,经氯化铵溶液
0.015);
23.优选地,所述3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酸酯、氨、甲醇钠 的摩尔比为3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酸酯:氨:甲醇钠 =1:4.62:0.01;
24.优选地,所述搅拌的时间为25-35min;所述加热回流反应的时间为4.5-5.5h; 所述氨解反应为在氮气环境下加热回流5.5-6.5h;
25.当(b)reformatsky反应后氨解优选的参数为上述参数时,在保证制备的收 率、纯度最优的情况下还能保证良好的经济效益。
26.作为本发明所述合成方法的优选实施方式,所述步骤(1)中,(c)reformatsky 反应具体包括以下步骤:将锌粉和卤代试剂加入到有机溶液中并于氮气环境下 搅拌,接着往里滴加2,5-二甲氧基苯甲醛溶解在有机溶剂里的混合溶液,滴加结 束后进行加热回流反应,反应结束后,经氯化铵溶液水解、乙酸乙酯萃取并重 结晶,得3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺;所述卤代试剂包括2
‑ꢀ
氯丙酰胺、2-溴丙酰胺或2-碘丙酰胺;所述有机溶剂包括四氢呋喃、2-甲基四氢 呋喃、甲苯或二甲苯;
27.优选地,所述2,5-二甲氧基苯甲醛、卤代试剂和锌粉的摩尔比为2,5-二甲氧 基苯甲醛:卤代试剂:锌粉=1:(1.0-1.4):(0.1-0.4);
28.优选地,所述2,5-二甲氧基苯甲醛、卤代试剂和锌粉的摩尔比为2,5-二甲氧 基苯甲醛:卤代试剂:锌粉=1:1.1:0.2;
29.优选地,所述搅拌的时间为25-35min;所述加热回流反应的时间为4.5-5.5h。
30.作为本发明所述合成方法的优选实施方式,所述步骤(2)中,霍夫曼降解 反应的具体包括以下步骤:控制温度在5℃以下将3-(2,5-二甲氧基苯基)-3-羟 基-2-甲基丙酰胺加入到次卤酸钠的氢氧化钠溶液中反应,接着将反应体系温度 升温至80-90℃下继续反应,反应结束后冷却至室温,抽滤,得甲氧明;所述次 卤酸钠为次溴酸钠或次氯酸钠;
31.优选地,所述次卤酸钠的氢氧化钠溶液为现配现用,具体配制方法为控制 温度在5℃以下将卤素加入到氢氧化钠溶液中搅拌25-30min;所述卤素为溴素或 氯气;所述氢氧化钠的质量百分数为30%;
32.优选地,控制温度在5℃以下的反应时间为50-70min,将反应体系温度升温 至80-90℃下继续反应的时间为50-70min;
33.当(c)reformatsky反应优选的参数为上述参数时,在保证制备的收率、纯 度最优的情况下还能保证良好的经济效益。
34.作为本发明所述合成方法的优选实施方式,所述步骤(2)中,成盐具体包 括以下步骤:将甲氧明用无水乙醇溶解,控制温度在30℃以下滴加浓盐酸至体 系ph值为1-2,收集析出的固体并用无水乙醇重结晶得盐酸甲氧明;
35.与现有技术相比,本发明的有益效果为:
36.第一:本发明的技术方案采用简单易得的2,5-二甲氧基苯甲醛为起始原料, 可从三条反应路线中任意选择一条合成关键中间体3-(2,5-二甲氧基苯基)-3
‑ꢀ
羟基-2-甲基丙酰胺,再对关键中间体进行霍夫曼降解后成盐即得盐酸甲氧明, 提供的反应条件温和,反应过程中不采用催化氢化还原,不采用具有腐蚀性的 氯化氢气体,因此,反应过程整体安全可控,能够用于放大生产;
37.第二:本发明提供的技术方案生成的产物的纯度高,纯度在99.71%以上, 且没有
引入重金属和亚硝胺类杂质,降低了用药风险,能实际应用于生产。
附图说明
38.图1为3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈的1h nmr图谱;
39.图2为3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺的1h nmr图谱;
40.图3为3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酸甲酯的1h nmr图谱;
41.图4为盐酸甲氧明的1h nmr图谱。
具体实施方式
42.为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对 本发明作进一步说明。
[0043][0044]
盐酸甲氧明的合成路线如上反应式所示,除另外说明外,反应中的化合 物都是通过常规购买途径得到;其中,反应式中的x代表氯、溴或碘,n为 0或1。
[0045]
实施例1
[0046]
本发明实施例的盐酸甲氧明通过(a)途径制备得到,合成的总收率为 52.40%,纯度为99.84%,具体合成方法如下:
[0047]
(1)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈的合成:
[0048]
往250ml三口瓶中加入镁屑(1.93g,79.52mmol)、100ml干燥的四氢 呋喃,再向其中加入2-溴丙腈(10.10g,75.90mmol)和3滴碘甲烷(约为 0.033mmol),氮气保护下保持回流反应1h后,降温至室温,向其中滴加溶 解了2,5-二甲氧基苯甲醛(12.00g,72.29mmol)的60ml干燥四氢呋喃溶液, 室温下反应1h后,加入氯化铵溶液水解,乙酸乙酯萃取、减压浓缩,得目标 产物3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈14.75g,收率92.3%;
[0049]
核磁表征:1h nmr(500mhz,dmso):6.92~7.12(2h,m),6.84~6.87(1h, m)6.74~6.77(1h,m),5.24~5.29(1h,m),3.73(3h,s),3.70(3h,s), 1.15~1.19(1h,m),0.84(3h,d,j=7.6)。
[0050]
(2)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺的合成:
[0051]
往250ml三口瓶中加入3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈(14.00g, 63.35mmol)、70ml二甲基亚砜溶液、碳酸钾(4.38g,31.68mmol),将混合 物控温30℃以下滴加质量百分数为35%的过氧化氢溶液(5.72ml, 66.52mmol),加毕反应3h后,将反应液倒入
300ml水中析出固体,抽滤后 收集固体并用水洗涤,再经乙酸乙酯重结晶后得目标产物3-(2,5-二甲氧基苯 基)-3-羟基-2-甲基丙酰胺12.48g,收率82.4%;
[0052]
核磁表征:1h nmr(500mhz,dmso):7.73(2h,br),6.91~7.10(2h,m), 6.84~6.87(1h,m)6.74~6.76(1h,m),5.25~5.28(1h,m),3.73(3h,s),3.70(3h, s),1.16~1.20(1h,m),0.85(3h,d,j=7.3)。
[0053]
(3)盐酸甲氧明的合成:
[0054]
控制温度在0℃以下向质量百分数为30%氢氧化钠溶液(60ml)中滴加 溴素(8.00g,50.18mmol),加毕继续搅拌30min,再向其中加入3-(2,5-二甲 氧基苯基)-3-羟基-2-甲基丙酰胺(10.00g,41.82mmol),加毕继续反应1h, 接着升温至80-90℃下反应1h,降至室温后抽滤得甲氧明固体,固体用无水 乙醇溶解后控温30℃以下滴加浓盐酸调节ph至1-2析出盐酸甲氧明粗品, 粗品用无水乙醇重结晶得盐酸甲氧明精制品7.13g,收率68.9%;
[0055]
核磁表征:1h nmr(500mhz,dmso):8.28(3h,s),7.00(1h,d,j=3.0) 6.90(1h,d,j=9.0),6.81(1h,dd,j=3.0,9.0),5.94(1h,d,j=4.8),5.12~5.16(1h, m),3.73(3h,s),3.69(3h,s),3.36~3.42(1h,m),0.90(3h,d,j=6.8)。
[0056]
实施例2
[0057]
本发明实施例的盐酸甲氧明通过(b)途径制备得到,合成的总收率为 40.76%,纯度为99.90%,具体合成方法如下:
[0058]
(1)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酸甲酯的合成:
[0059]
往250ml三口瓶中加入活化好的锌粉(7.88g,12.05mmol)、150ml干 燥的四氢呋喃、2-溴丙酸甲酯(10.50g,63.25mmol),氮气保护下室温搅拌 30min后,向其中滴加溶解了2,5-二甲氧基苯甲醛(10.00g,60.24mmol)的 50ml干燥四氢呋喃溶液,氮气保护下回流反应5h,降温至室温后经氯化铵 溶液水解,乙酸乙酯萃取蒸干后得目标产物3-(2,5-二甲氧基苯基)-3-羟基-2-甲 基丙酸甲酯11.11g,收率72.6%;
[0060]
核磁表征:1h nmr(500mhz,dmso):6.93~7.11(2h,m),6.85~6.88(1h, m)6.75~6.78(1h,m),5.28~5.30(1h,m),3.73(3h,s),3.70(3h,s),3.60(3h,s), 1.23~1.27(1h,m),0.84(3h,d,j=7.1)。
[0061]
(2)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺的合成:
[0062]
往250ml三口瓶中加入3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酸甲酯 (11.00g,43.29mmol),再向其中加入2mol/l氨的甲醇溶液(100ml)、 甲醇钠(0.02g,0.43mmol),氮气保护下回流反应6h,蒸除溶剂后,向剩 余物中加入水搅拌,抽滤水洗后,经乙酸乙酯重结晶得目标产物3-(2,5-二甲 氧基苯基)-3-羟基-2-甲基丙酰胺8.67g,收率83.8%;
[0063]
(3)盐酸甲氧明的合成:
[0064]
控制温度在0℃以下向质量百分数为30%氢氧化钠溶液(50ml)中滴加 溴素(6.40g,40.14mmol),加毕继续搅拌30min,再向其中加入3-(2,5-二甲 氧基苯基)-3-羟基-2-甲基丙酰胺(8.00g,33.46mmol),加毕继续反应1h,接 着升温至80-90℃下反应1h,降至室温后抽滤得甲氧明固体,固体用无水乙 醇溶解后控温30℃以下滴加浓盐酸调节ph至1-2析出盐酸甲氧明粗品,粗 品用无水乙醇重结晶得盐酸甲氧明精制品5.55g,收率67.0%。
[0065]
实施例3
[0066]
本发明实施例的盐酸甲氧明通过(c)途径制备得到,合成的总收率为 43.66%,纯度为99.74%,具体合成方法如下:
[0067]
(1)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺的合成:
[0068]
往250ml三口瓶中加入活化好的锌粉(7.88g,12.05mmol)、150ml干 燥的四氢呋喃、2-溴丙酰胺(10.01g,66.27mmol),氮气保护下室温搅拌30min 后,向其中滴加溶解了2,5-二甲氧基苯甲醛(10.00g,60.24mmol)的50ml 干燥四氢呋喃溶液,氮气保护下回流反应5h,降温至室温后经氯化铵溶液水 解,乙酸乙酯萃取、减压浓缩,再用乙酸乙酯重结晶后得目标产物3-(2,5-二 甲氧基苯基)-3-羟基-2-甲基丙酰胺9.27g,收率64.4%;
[0069]
(3)盐酸甲氧明的合成:
[0070]
控制温度在0℃以下向质量百分数为30%氢氧化钠溶液(60ml)中滴加 溴素(7.20g,45.16mmol),加毕继续搅拌30min,再向其中加入3-(2,5-二甲 氧基苯基)-3-羟基-2-甲基丙酰胺(9.00g,37.64mmol),加毕继续反应1h,接 着升温至80-90℃下反应1h,降至室温后抽滤得甲氧明固体,固体用无水乙 醇溶解后控温30℃以下滴加浓盐酸调节ph至1-2析出盐酸甲氧明粗品,粗 品用无水乙醇重结晶得盐酸甲氧明精制品6.31g,收率67.8%。
[0071]
实施例4
[0072]
本发明实施例的盐酸甲氧明通过(a)途径制备得到,合成的总收率为 45.68%,纯度为99.79%,其与实施例1不同的地方在于改变了3-(2,5-二甲氧基 苯基)-3-羟基-2-甲基丙腈的合成过程中2,5-二甲氧基苯甲醛、金属催化剂、卤代 试剂和碘甲烷的摩尔比,同时改变了3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺 的合成过程中3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈、碱和过氧化氢的摩 尔比;
[0073]
具体合成方法如下:
[0074]
(1)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈的合成:
[0075]
往250ml三口瓶中加入镁屑(1.75g,72.29mmol)、100ml干燥的四氢 呋喃,再向其中加入2-溴丙腈(9.62g,72.29mmol)和3滴碘甲烷(约为 0.033mmol),氮气保护下保持回流反应1h后,降温至室温,向其中滴加溶 解了2,5-二甲氧基苯甲醛(12.00g,72.29mmol)的60ml干燥四氢呋喃溶液, 室温下反应1h后,加入氯化铵溶液水解,乙酸乙酯萃取、减压浓缩,得目标 产物3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈13.58g,收率85.0%;
[0076]
(2)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺的合成:
[0077]
往250ml三口瓶中加入3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈(13.00g, 58.83mmol)、70ml二甲基亚砜溶液、碳酸钾(2.44g,17.65mmol),将混合 物控温30℃以下滴加质量百分数为35%的过氧化氢溶液(5.06ml, 58.83mmol),加毕反应3h后,将反应液倒入300ml水中析出固体,抽滤后 收集固体并用水洗涤,再经乙酸乙酯重结晶后得目标产物3-(2,5-二甲氧基苯 基)-3-羟基-2-甲基丙酰胺10.97g,收率78.0%;
[0078]
(3)盐酸甲氧明的合成:
[0079]
控制温度在0℃以下向质量百分数为30%氢氧化钠溶液(60ml)中滴加 溴素(8.00g,50.18mmol),加毕继续搅拌30min,再向其中加入3-(2,5-二甲 氧基苯基)-3-羟基-2-甲基丙酰胺(10.00g,41.82mmol),加毕继续反应1h, 接着升温至80-90℃下反应1h,降至室温后抽滤得甲氧明固体,固体用无水 乙醇溶解后控温30℃以下滴加浓盐酸调节ph至1-2析出盐酸甲氧明粗品, 粗品用无水乙醇重结晶得盐酸甲氧明精制品7.13g,收率68.9%。
[0080]
实施例5
[0081]
本发明实施例的盐酸甲氧明通过(a)途径制备得到,合成的总收率为 44.89%,纯度为99.71%,其与实施例1不同的地方在于改变了3-(2,5-二甲氧基 苯基)-3-羟基-2-甲基丙腈的合成过程中的卤代试剂,本实施例采用2-氯丙腈, 同时改变了3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺的合成过程中所用的 碱,本实施例采用叔丁醇钠;
[0082]
具体合成方法如下:
[0083]
(1)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈的合成:
[0084]
往250ml三口瓶中加入镁屑(1.93g,79.52mmol)、100ml干燥的四氢 呋喃,再向其中加入2-氯丙腈(6.79g,75.90mmol)和3滴碘甲烷(约为 0.033mmol),氮气保护下保持回流反应1h后,降温至室温,向其中滴加溶 解了2,5-二甲氧基苯甲醛(12.00g,72.29mmol)的60ml干燥四氢呋喃溶液, 室温下反应1h后,加入氯化铵溶液水解,乙酸乙酯萃取、减压浓缩,得目标 产物3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈14.01g,收率87.7%;
[0085]
(2)3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙酰胺的合成:
[0086]
往250ml三口瓶中加入3-(2,5-二甲氧基苯基)-3-羟基-2-甲基丙腈(14.00g, 63.35mmol)、70ml二甲基亚砜溶液、叔丁醇钠(3.04g,31.68mmol),将混 合物控温30℃以下滴加质量百分数为35%的过氧化氢溶液(5.72ml, 66.52mmol),加毕反应3h后,将反应液倒入300ml水中析出固体,抽滤后 收集固体并用水洗涤,再经乙酸乙酯重结晶后得目标产物3-(2,5-二甲氧基苯 基)-3-羟基-2-甲基丙酰胺11.26g,收率74.3%;
[0087]
(3)盐酸甲氧明的合成:
[0088]
控制温度在0℃以下向质量百分数为30%氢氧化钠溶液(60ml)中滴加 溴素(8.00g,50.18mmol),加毕继续搅拌30min,再向其中加入3-(2,5-二甲 氧基苯基)-3-羟基-2-甲基丙酰胺(10.00g,41.82mmol),加毕继续反应1h, 接着升温至80-90℃下反应1h,降至室温后抽滤得甲氧明固体,固体用无水 乙醇溶解后控温30℃以下滴加浓盐酸调节ph至1-2析出盐酸甲氧明粗品, 粗品用无水乙醇重结晶得盐酸甲氧明精制品7.13g,收率68.9%。
[0089]
从实施例1-3可以看出,采用本发明提供的技术方案合成得到的盐酸甲 氧明收率高,其中采用(a)路线制备得到的收率可达52.40%,纯度可达 99.84%;从实施例1和实施例4-5可以看出,采用(a)路线制备时,反应物 的摩尔比以及对试剂的选择都会对合成收率产生影响。
[0090]
最后应当说明的是,以上实施例以说明本发明的技术方案而非对本发明保 护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技 术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离 本发明技术方案的实质和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。