大规模制备2,4,6-三氟-n-[6-(1-甲基-哌啶-4-羰基)-吡啶-2-基]-苯甲酰胺半琥珀酸盐和制备2,4,6-三氟-n-[6-(1-甲基-哌啶-4-羰基)-吡啶-2-基]-苯甲酰胺乙酸盐的方法和中间体
[0001]
本发明的实施方案涉及药物化学和合成有机化学领域,并提供了大规模合成2,4,6-三氟-n-[6-(1-甲基-哌啶-4-羰基)-吡啶-2-基]-苯甲酰胺半琥珀酸盐(5-ht1f受体激动剂)的方法和中间体,以及通过这些方法制备的制剂和产品形式,并涉及2,4,6-三氟-n-[6-(1-甲基-哌啶-4-羰基)-吡啶-2-基]-苯甲酰胺乙酸盐的制备及其用于肠胃外制剂和治疗偏头痛的用途。
[0002]
拉司米地坦(lasmiditan)是选择性和高效的5-ht
1f
受体激动剂,现在作为50mg或100mg片剂在美国被批准,用于紧急按需治疗偏头痛(参见,例如rubio-beltr
á
n等人,pharmacol ther 2018;186:88-97,和lasmiditan for the treatment of migraine,capi,m等人,expert opinion investigational drugs,(2017),第26卷,第2期,227-234)。拉司米地坦(col 144,ly 573144,cas登记号439239-90-4)在化学上可以描述为2,4,6-三氟-n-[6-(1-甲基-哌啶-4-基羰基)-吡啶-2-基]-苯甲酰胺。美国专利7,423,050和美国公开20080300407描述了具有以下结构式的2,4,6-三氟-n-[6-(1-甲基-哌啶-4-羰基)-吡啶-2-基]-苯甲酰胺的半琥珀酸盐:
[0003][0004]
制备拉司米地坦和盐及其某些多晶型形式、制剂和剂型的方法是本领域技术人员已知的,并且描述于例如wo 2003/084949、wo 2011/123654和wo 2018/106657中。
[0005]
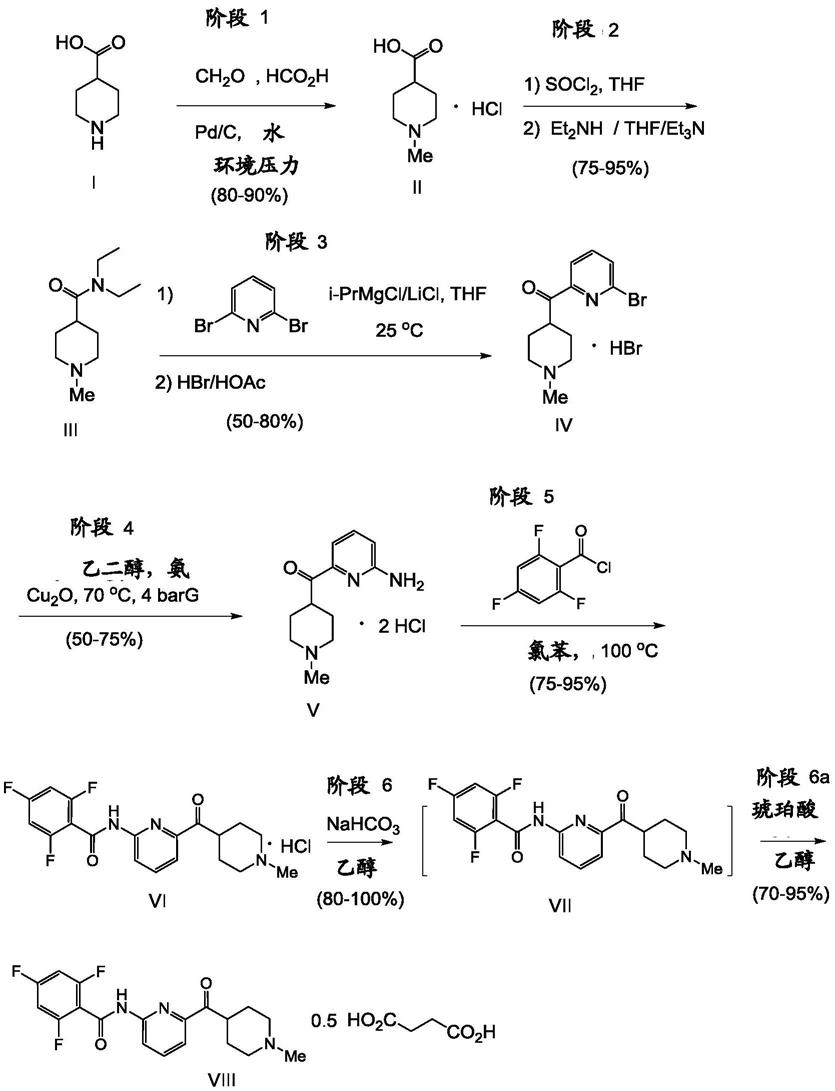
如本文所用,拉司米地坦的有用形式包括其药学上可接受的盐,包括但不限于2,4,6-三氟-n-[6-(1-甲基-哌啶-4-基羰基)-吡啶-2-基]-苯甲酰胺单盐酸盐,和2,4,6-三氟-n-[6-(1-甲基-哌啶-4-羰基)-吡啶-2-基]-苯甲酰胺半琥珀酸盐。制备2,4,6-三氟-n-[6-(1-甲基-哌啶-4-羰基)-吡啶-2-基]-苯甲酰胺的半琥珀酸盐的合成路线已经如以下流程a中所示在先前已公开。从市售哌啶4-甲酸开始,通过下面流程a中描述的路线,在所有9个步骤中,拉司米地的总收率为约10-46%。改进拉司米地坦的合成可以提供大量和多样的益处,特别是对于大规模生产。
[0006]
流程a
[0007][0008]
合成化学工艺路线可以重新设计或修改,目的是实现各种优点,包括例如:提高收率,获得结晶产物,降低杂质分布,利用可商购的中间体,使所需的合成步骤的数量最小化,降低所需的输入量和/或产生的副产物,或这些改进的任何有用组合,以实现重要的现实结果,包括降低成本,提供资源密集较少的方法,和促进有效生产。需要改进的制备拉司米地坦的方法,其可以实现一个或多个这些目的,特别是对于大规模合成。
[0009]
此外,偏头痛是急诊室中最常见的表现症状之一。当对由于恶心和/或呕吐而难以施用片剂的患者使用拉司米地坦时,目前在急诊室环境中缓解头痛的方法可能需要依赖于制备约1mg/ml拉司米地坦的稀释制剂,其在延长的时间内例如约20-60分钟内静脉内递送。在临床研究中,拉司米地坦以约1-60mg的剂量静脉内递送,该剂量在20分钟内以60ml输注
递送(参见美国专利申请公开号2010/0256187)。对于不能施用片剂的患者,用拉司米地坦安全有效地治疗偏头痛将通过高浓度的肠胃外剂型的可获得性来成为可能。本公开还解决了这种需要。
[0010]
发明概述
[0011]
本发明的实施方案提供了制备拉司米地坦半琥珀酸盐(2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺半琥珀酸盐)和/或其组合物的方法,和/或用于这些方法的特别有用的中间体。本发明的实施方案进一步提供了拉司米地坦乙酸盐(2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺乙酸盐)和/或其组合物的制备,和/或拉司米地坦乙酸盐及其制剂在皮下药物递送中的用途。
[0012]
在一个实施方案中,本发明提供了一种称为路线i的制备下式化合物的方法:
[0013][0014]
包括以下步骤:
[0015]
i.)在还原胺化条件下处理哌啶-4-甲酸,所述还原胺化条件包括在水中的甲醛和甲酸,随后用hcl水溶液处理,接着水蒸馏以及加入乙腈,重复稀释/蒸馏直到通过karl-fischer分析水含量不超过0.2%,得到固体1-甲基哌啶-4-甲酸盐酸盐;
[0016]
ii.)用氯化剂如亚硫酰氯在氯苯中处理1-甲基哌啶-4-甲酸盐酸盐,得到1-甲基哌啶-4-甲酰氯;
[0017]
iii.)用n,n-二乙胺在含有三乙胺的氯苯中处理1-甲基哌啶-4-甲酰氯,随后碱洗,随后用在异丙醇中的hcl水溶液处理,得到固体n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合物盐酸盐;
[0018]
iv.)在非极性溶剂如甲基叔丁基醚中,用无机碱如naoh水溶液处理n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合盐酸盐,随后水洗、相分离,并蒸馏有机溶剂,直至通过karl fischer分析测得水含量不超过0.1重量%,得到n,n-二乙基-1-甲基-哌啶-4-甲酰胺;
[0019]
v.)随后在非极性有机溶剂如甲基叔丁基醚中用(6-溴-2-吡啶基)锂处理n,n-二乙基-1-甲基-哌啶-4-甲酰胺,随后用水和合适的有机溶剂如正丁醇萃取所得混合物,相分离,并重复蒸馏有机溶剂直到通过karl-fischer分析水含量不超过0.2重量%,得到(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮;
[0020]
vi.)用hbr水溶液处理(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮,随后用正丁醇萃取,然后重复蒸馏有机溶剂,直到通过karl-fischer分析水含量不超过0.3%,得到固体(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮氢溴酸盐;
[0021]
vii.)在cu2o催化剂的存在下,在约80℃,用nh3在乙二醇中的溶液处理(6-溴-2-吡啶基-1-甲基-4-哌啶基)甲酮氢溴酸盐约2小时,随后用水、饱和nacl水溶液和20%naoh水溶液洗涤,随后用非极性非质子溶剂如甲基叔丁基醚提取,相分离,并用5重量%碳处理有
机相;
[0022]
viii.)过滤上述混合物,用合适的极性醇溶剂如异丙醇稀释,重复蒸馏有机溶剂直到通过karl-fischer分析水含量不超过0.2%,随后用异丙醇、水和20重量%hcl处理得到的残余物,其中得到的浆液的水浓度至少为2%,过滤得到的浆液,在40℃真空干燥16-24小时,得到固体(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐;
[0023]
ix.)在氯苯中用6重量/重量%naoh水溶液在约54℃处理(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐约30分钟,随后相分离,并真空蒸馏水溶液,得到(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮;
[0024]
x.)随后在约100℃,在氯苯中用2,4,6-三氟苯甲酰氯处理(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮约4小时,随后冷却,加入乙腈并将所得浆液加热至80℃约1小时,随后通过过滤收集所得固体,得到固体2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺盐酸盐;
[0025]
xi.)在甲基叔丁基醚中用饱和的na2co3水溶液处理2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺盐酸盐;
[0026]
xii.)用sio2处理上述步骤xi的混合物,随后过滤,用碳处理,过滤,以及蒸发,用乙醇稀释,并蒸馏直到通过karl-fischer分析水含量不超过1%,得到2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺;
[0027]
xiii.)在约55℃将在乙醇中的2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺用0.5当量琥珀酸的乙醇溶液处理,在室温不少于3小时,随后通过滤收集固体,得到固体2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺半琥珀酸盐。
[0028]
在上述路线i的方法中,优选使用分批处理方法进行反应。在一个实施方案中,路线i的批次以工艺规模生产。在一个实施方案中,路线i的批次以至少1千克生产。在一个实施方案中,路线i的批次以至少10千克生产。在一个实施方案中,路线i的批次以至少100千克生产。
[0029]
在上述路线i的方法中,氯苯的使用避免了在其它方法中发生的降解,如thf,其与酰氯在规模(例如100kg)下反应,导致基本上没有酰氯的产量。
[0030]
在另一个实施方案中,本发明提供了一种称为路线ii的制备下式化合物的方法:
[0031][0032]
包括以下步骤:
[0033]
i.)在还原胺化条件下处理哌啶-4-甲酸,所述还原胺化条件包括在水中的甲醛和甲酸,随后用hcl水溶液处理,接着水蒸馏以及加入乙腈,重复稀释/蒸馏直到通过karl-fischer分析水含量不超过0.2%,得到固体1-甲基哌啶-4-甲酸盐酸盐;
[0034]
ii.)用氯化剂如亚硫酰氯在氯苯中处理1-甲基哌啶-4-甲酸盐酸盐,得到1-甲基
哌啶-4-甲酰氯;
[0035]
iii.)用n,n-二乙胺在含有三乙胺的氯苯中处理1-甲基哌啶-4-甲酰氯,随后碱洗,随后用在异丙醇中的hcl水溶液处理,得到固体n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合物盐酸盐;
[0036]
iv.)在非极性溶剂如甲基叔丁基醚中,用无机碱如naoh水溶液处理n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合物盐酸盐,随后水洗、相分离,并蒸馏有机溶剂,直至通过karl fischer分析测得水含量不超过0.1重量%,得到n,n-二乙基-1-甲基-哌啶-4-甲酰胺;
[0037]
v.)随后在非极性有机溶剂如甲基叔丁基醚中用(6-溴-2-吡啶基)锂处理n,n-二乙基-1-甲基-哌啶-4-甲酰胺,随后用水和合适的有机溶剂如正丁醇萃取所得混合物,相分离,并重复蒸馏有机溶剂直到通过karl-fischer分析水含量不超过0.2重量%,得到(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮;
[0038]
vi.)用hbr水溶液处理(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮,随后用正丁醇萃取,然后重复蒸馏有机溶剂,直到通过karl-fischer分析水含量不超过0.3%,得到固体(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮氢溴酸盐;
[0039]
vii.)在水和甲苯的两相混合物中用固体koh处理(6-溴-2-吡啶基-1-甲基-4-哌啶基)甲酮氢溴酸盐约3小时,随后分离有机层并蒸发溶剂,得到(6-溴-2-吡啶基-1-甲基-4-哌啶基)甲酮;
[0040]
viii.)在含有k2co3、水、pd(oac)2和xantphos的甲苯中,在约70℃用2,4,6-三氟苯甲酰胺处理(6-溴-2-吡啶基-1-甲基-4-哌啶基)甲酮约12小时,直到通过hplc测定(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮的含量不超过0.1%,随后用水和etoac稀释该反应混合物,随后在60℃用硫脲改性的硅胶处理约8小时,接着过滤,得到2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺的溶液;
[0041]
ix.)在55℃将2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺在etoac的溶液用约0.5当量琥珀酸溶于etoh中的溶液处理约3小时,随后在约10小时内冷却至室温,过滤收集得到的固体,得到固体2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺半琥珀酸盐。
[0042]
在上述路线ii的方法中,优选使用分批处理方法进行反应。在一个实施方案中,路线ii的批次以工艺规模生产。在一个实施方案中,路线ii的批次以至少1千克生产。在一个实施方案中,路线ii的批次以至少10千克生产。在一个实施方案中,路线ii的批次以至少100千克生产。
[0043]
在上述路线ii的方法中,氯苯的使用避免了在其它方法中发生的降解,如thf,其与酰氯在规模(例如100kg)下反应,导致基本上没有酰氯的产量。
[0044]
在另一个实施方案中,本发明提供了:
[0045][0046]
其可被命名为(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐。优选地,该化合物是结晶的。(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐特别适用于制备2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺半琥珀酸盐,且使用(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐的方法可以提供有利的方法特征,包括但不限于中间体和/或最终材料的纯度。(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐被认为是6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮的一种新的稳定水合形式。本文所述的分离(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐的方法提供了改进的杂质脱除和改进的受控结晶方法。各形态和化学稳定性研究表明(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐通常是稳定的,各干燥研究表明过度干燥以除去水合水是困难的,即使在加强条件下也是如此。使用该中间体有利地以预期收率提供高纯度产物。
[0047]
在另一个实施方案中,本公开提供了拉司米地坦乙酸盐,其可以由下式表示:
[0048][0049]
在另一个实施方案中,本发明提供结晶形式的拉司米地坦乙酸盐,并且进一步提供了如下的结晶形式的拉司米地坦乙酸盐,其特征在于使用cukα辐照的x射线粉末衍射图,所述x射线粉末衍射图具有在衍射角2θ26.2
°
处的强峰,以及选自20.4
°
、14.0
°
和17.9
°
(分别
±
0.2
°
)的一个或多个峰。在另一个实施方案中,本发明提供了一种药物组合物,其包含根据上述实施方案的拉司米地坦乙酸盐和一种或多种药学上可接受的载体、稀释剂或赋形剂。优选地,药物组合物包含乙酸。优选地,药物组合物包含乙酸并且用于皮下施用。
[0050]
在另一个实施方案中,本发明提供了治疗患者偏头痛的方法,所述方法包括向需要这种治疗的患者施用有效量的拉司米地坦乙酸盐。在另一个实施方案中,本发明提供了治疗患者偏头痛的方法,所述方法包括向需要这种治疗的患者施用有效量的拉司米地坦乙酸盐与一种或多种药学上可接受的载体、稀释剂或赋形剂。在另一个实施方案中,本发明提供了治疗患者偏头痛的方法,所述方法包括向需要这种治疗的患者施用有效量的拉司米地坦乙酸盐与一种或多种药学上可接受的载体、稀释剂或赋形剂,其中所述组合物包含乙酸。
[0051]
在另一个实施方案中,本发明提供了用于治疗法的拉司米地坦乙酸盐。在另一个实施方案中,本发明提供了用于治疗法的拉司米地坦乙酸盐与一种或多种药学上可接受的
载体、稀释剂或赋形剂的药物组合物。在另一个实施方案中,本发明提供了一种用于治疗法的拉司米地坦乙酸盐和一种或多种药学上可接受的载体、稀释剂或赋形剂的药物组合物,其中所述组合物包含乙酸。
[0052]
在另一个实施方案中,本发明提供了用于治疗偏头痛的拉司米地坦乙酸盐。在另一个实施方案中,本发明提供了用于治疗偏头痛的拉司米地坦乙酸盐与一种或多种药学上可接受的载体、稀释剂或赋形剂的药物组合物。在另一个实施方案中,本发明提供了一种用于治疗偏头痛的拉司米地坦乙酸盐与一种或多种药学上可接受的载体、稀释剂或赋形剂的药物组合物,其中所述组合物含有乙酸。
[0053]
在另一个实施方案中,本公开提供了拉司米地坦乙酸盐,以及在水性载体中包含高浓度的拉司米地坦乙酸盐(例如约10-200mg/ml游离碱当量(free-base equivalent))的药物组合物。在实施方案中,所述药物组合物包含在缓冲含水溶液中的约10-200mg/ml游离碱当量的拉司米地坦。在实施方案中,所述缓冲含水溶液在37℃时的ph为ph 6.0-7.5。在实施方案中,所述缓冲含水溶液包含乙酸。
[0054]
除了水性载体(优选无菌的去离子蒸馏水)之外,本文所述的药物组合物还可以包含一种或多种药学上可接受的赋形剂或助溶剂。术语“药学上可接受的”是指赋形剂和助溶剂,其适用于与人类组织接触而没有过度的毒性、刺激、过敏反应或其它问题或并发症,与合理的利益/风险比相称。药物组合物及其制备方法是本领域公知的(参见,例如remington:the scienceand practice of pharmacy(a.gennaro等编,第21版,mack publishing co.,2005))。
[0055]
可以以散装或以剂量单位形式提供拉司米地坦乙酸盐的药物组合物。特别有利的是,配制单位剂量形式的拉司米地坦乙酸盐的药物组合物,以便于施用和剂量的一致性。如本文所用的术语“剂量单位形式”是指适合作为用于待治疗的个体的单位剂量的物理上离散的单位;每个单位含有经计算产生所需的治疗效果的预定量的活性化合物拉司米地坦与所需的药物载体。剂量单位形式可以是例如安瓿、小瓶或注射器。
[0056]
在实施方案中,本公开提供了包含一定量的如本文所述的拉司米地坦乙酸盐的药物组合物,其中所述量为每剂量10mg至200mg。在实施方案中,本公开提供了包含一定量的如本文所述的拉司米地坦乙酸盐的药物组合物,其中所述量为每剂量10mg、20mg、30mg、40mg、50mg、100mg或200mg。上述剂量基于平均体重的成人。对于体重较轻的个体,例如老人或儿童,较小的剂量是可接受的。因此,在实施方案中,药物组合物可以包含较小的剂量,例如5、10或15mg。
[0057]
如本文所述,所述高浓度的拉司米地坦乙酸盐含水溶液使得能够通过注射高浓度拉司米地坦水溶液(例如通过静脉内、皮下或肌内途径)施用单一治疗有效剂量。
[0058]
在实施方案中,本公开提供了高浓度的拉司米地坦乙酸盐含水溶液。在实施方案中,所述高浓度的拉司米地坦乙酸盐含水溶液被配制成肠胃外剂型。在实施方案中,所述高浓度含水溶液含有10-200mg/ml游离碱当量的拉司米地坦。在实施方案中,所述高浓度的拉司米地坦乙酸盐含水溶液是肠胃外剂型的形式。在实施方案中,所述肠胃外剂型是10-200mg/ml游离碱当量的拉司米地坦的缓冲含水溶液。在实施方案中,所述肠胃外剂型是10、20、30、40、50、100或200mg/ml游离碱当量的拉司米地坦的缓冲含水溶液。在实施方案中,所述肠胃外剂型适合于皮下或肌内注射。优选地,所述肠胃外剂型用于皮下注射。在实施方案
中,缓冲溶液的ph在37℃在ph 6.0-7.5之间。
[0059]
在实施方案中,缓冲含水溶液包含基于有机酸的缓冲体系。在实施方案中,有机酸是二羧酸或三羧酸。在实施方案中,所述二羧酸或三羧酸选自乙酸和柠檬酸。在实施方案中,所述有机酸是琥珀酸。在实施方案中,所述缓冲液是乙酸缓冲液。在实施方案中,所述缓冲含水溶液不含有机溶剂。在实施方案中,所述缓冲含水溶液不含有机溶剂和表面活性剂。在优选的实施方案中,所述缓冲含水溶液包含拉司米地坦乙酸盐、及乙酸和氢氧化钠,在37℃下将ph调节至6.0-7.5。
[0060]
在实施方案中,拉司米地坦乙酸盐的肠胃外剂型以适于通过皮下途径施用的预填充注射器的形式提供。在实施方案中,预填充的注射器包含10-50mg/ml游离碱当量的拉司米地坦。在实施方案中,预填充的注射器包含10、20、30、40、50或100mg/ml游离碱当量的拉司米地坦。在实施方案中,在缓冲含水溶液中提供拉司米地坦,所述缓冲含水溶液在37℃具有ph6.0-7.5。在实施方案中,预填充注射器适于家庭使用,例如,用于可能面临头痛极端和快速发作的那些偏头痛患者。在实施方案中,预填充注射器包含在具有用于肠胃外施用、优选皮下注射施用的说明书的包装中。在实施方案中,预填充的注射器是具有用于皮下注射的说明书的自动注射器的形式。
[0061]
在实施方案中,拉司米地坦乙酸盐的肠胃外剂型以含有10-50mg/ml游离碱当量的拉司米地坦的小瓶的形式提供。在实施方案中,拉司米地坦乙酸盐的肠胃外剂型以含有10、20、30、40、50或100mg/ml游离碱当量的拉司米地坦的小瓶的形式提供。在实施方案中,在缓冲含水溶液中提供拉司米地坦,所述缓冲含水溶液在37℃具有ph 6.0-7.5。
[0062]
本公开还提供了用于偏头痛发作的急性治疗的方法,所述方法包括施用治疗有效剂量的如本文所述的拉司米地坦乙酸盐。在实施方案中,肠胃外溶液通过皮下注射施用。在实施方案中,肠胃外溶液在缓冲含水溶液中包含10-50mg/ml游离碱当量的拉司米地坦乙酸盐,所述缓冲含水溶液在37℃具有ph 6.0-7.5。在实施方案中,肠胃外溶液包含10、20、30、40、50或100mg/ml游离碱当量的拉司米地坦。在实施方案中,所述方法包括施用单一治疗有效剂量的少于或等于1ml、如约0.5至1ml体积的拉司米地坦乙酸盐,例如通过单次皮下注射施用。在实施方案中,注射体积为约1ml。在实施方案中,注射体积为约0.5ml。
[0063]
本发明提供了一种在有需要的患者中治疗偏头痛的方法,所述方法包括向患者施用每皮下剂量20-200mg的拉司米地坦乙酸盐和药学上可接受的稀释剂或载体。本发明提供了一种在有需要的患者中治疗偏头痛的方法,所述方法包括向患者施用每皮下剂量20mg的拉司米地坦乙酸盐和药学上可接受的稀释剂或载体。本发明提供了一种在有需要的患者中治疗偏头痛的方法,所述方法包括向患者施用每皮下剂量50mg的拉司米地坦乙酸盐和药学上可接受的稀释剂或载体。本发明提供了一种在有需要的患者中治疗偏头痛的方法,所述方法包括向患者施用每皮下剂量75mg的拉司米地坦乙酸盐和药学上可接受的稀释剂或载体。本发明提供一种在有需要的患者中治疗偏头痛的方法,其包括向患者施用每皮下剂量100mg的拉司米地坦乙酸盐和药学上可接受的稀释剂或载体。本发明提供了一种在有需要的患者中治疗偏头痛的方法,所述方法包括向患者施用每皮下剂量150mg的拉司米地坦乙酸盐和药学上可接受的稀释剂或载体。本发明提供一种在有需要的患者中治疗偏头痛的方法,所述方法包括向患者施用每皮下剂量200mg的拉司米地坦乙酸盐和药学上可接受的稀释剂或载体。
[0064]
在一些实施方案中,“患者”是已经被诊断为患有需要用本文所述的药物组合物预防的病症或障碍的人。在一些实施方案中,患者是特征在于处于病症或障碍的风险中的人,所述病症或障碍被指示了施用本文所述的药物组合物。在其中通过既定和可接受的分类已知的可以通过本发明方法治疗的障碍的情况下,例如偏头痛,发作性头痛,慢性头痛,慢性丛集性头痛和/或发作性丛集性头痛,其分类可以在多种来源中找到。例如,目前,第4版的diagnostic and statistical manual of mental disorders(dsm-ivtm)(1994,american psychiatric association,washington,d.c.)提供了诊断工具,用于鉴定本文所述的许多障碍。另外,international classification of diseases,tenth revision(icd-10)提供了本文所述许多障碍的分类。技术人员将认识到,存在本文所述障碍的替代命名法、疾病分类学和分类系统,包括dsm-iv和icd-10中描述的那些,并且术语和分类系统随着医学科学的发展而发展。可以进一步诊断偏头痛患者是否偏头痛伴有先兆(1.1和1.2),如international headache society(ihs)international classification of headache disorders,第三版,(ichd-3)beta version(the international classification of headache disorders,第三版(beta version),cephalalgia 2013;33:629-808)定义的。在一些实施方案中,人患者在接受拉司米地坦的长期施用(优选夜晚)来预防偏头痛之前已被诊断为患有发作性偏头痛。在一些实施方案中,人患者在接受抗体之前被诊断为患有慢性偏头痛。在一些实施方案中,人患者的偏头痛头痛出现先兆。在一些实施方案中,人患者的偏头痛头痛不出现先兆。
[0065]
如本文所用,“偏头痛”包括但不限于偏头痛发作。如本文所用,“偏头痛发作”是指以下描述。症状可能在偏头痛发作的各个阶段重叠,并且并非所有患者都经历相同的临床表现。在前驱症状阶段,大多数患者具有监测前症状,它们可能在头痛阶段之前长至72小时。这些包括情绪和活动的变化、易怒、疲劳、饮食冲动、反复打哈欠、颈强直和高声恐怖。这些症状可能会充分地持续到先兆、头痛、甚至是头痛后阶段。一些患者经历先兆阶段,其中约三分之一的患者在发作期间经历短暂的神经学缺陷。ichd-3将先兆定义为1种或多种短暂的、完全可逆的神经学缺陷,其中至少1种必须具有单侧定位,其发生历经5分钟或更长时间,并且其中每种缺陷持续5至60分钟。此时在超过90%的病例中发现了视觉先兆,其可能显示阳性(闪光暗点)、阴性(暗点)或这两种现象,并且最常见的缺陷,感觉、运动、言语、脑干和视网膜先兆症状也会出现。认为皮层神经元去极化的瞬态波是偏头痛先兆临床现象背后的病理生理学脑机制。在头痛阶段,头痛发作可能持续4至72小时,并且伴有恶心、畏光和高声恐怖,或两者兼有。头痛的特征在于单侧、搏动,中度或重度,并且因身体活动而加剧;这些特征中的两种足以满足诊断标准。在头痛后阶段,特征性症状反映出在监测前阶段观察到的症状。典型的头痛后症状包括疲倦、注意力不集中和颈强直。尚不清楚这些症状是否在监测前阶段开始并且在整个头痛阶段一直持续到头痛后阶段,是否也可能在头痛阶段开始,或者甚至在头痛阶段结束后出现。
[0066]
如本文所用,“偏头痛头痛”是指持续时间≥30分钟并且具有或不具有先兆的头痛,其中具有以下两个必需特征(a和b):a)以下头痛特征的至少两种:1)单侧定位,2)搏动性(pulsating quality),3)中度或重度疼痛强度,和4)因进行日常身体活动而加重或引起避免日常身体活动;和b)在头痛期间,存在以下情况的至少一种:a)恶心和/或呕吐,和/或b)畏光和高声恐怖。如本文所用,“可能的偏头痛头痛”是指持续时间大于30分钟的头痛,有
或没有先兆,但缺少international headache society ichd-3定义中的偏头痛特征之一。
[0067]
术语“有效量”或“治疗有效量”是指拉司米地坦乙酸盐在药物组合物中的量或剂量,例如施用中所施用的总剂量,其在单剂量或多剂量施用于患者时,提供给患者期望的药理作用,例如能够活化5-ht
1f
受体的量。在优选的实施方案中,“有效量”是指拉司米地坦乙酸盐的量,其在急性施用后能够在施用后使患者无偏头痛发作。“剂量”是指经计算可在患者中产生期望的治疗效果的拉司米地坦乙酸盐的预定量。如本文所用,“mg”是指毫克。如本文所用,以mg表示的剂量是指活性药物成分拉司米地坦的游离碱质量当量,例如“100mg”剂量是指作为游离碱当量的100mg活性药物成分拉司米地坦。如本文所用,一个给定剂量可以解释为描述了大约所示的量的剂量,即比所示的剂量高或低至多百分之十的剂量同样应被理解为以类似于所示的剂量的方式提供有用的方案。
[0068]
附图简述
[0069]
图1:含有马来酸(内标)的拉司米地坦乙酸盐的1h nmr谱(400mhz,dmso-d6)的图示。
[0070]
发明详述
[0071]
本文所述的反应可以通过本领域技术人员已知的标准技术通过使用常规玻璃器具进行,或者可以在设计用于这种转化的设备中以预初试验和/或工艺规模进行。此外,所描述的这些反应中的每一个都可以通过分批方法或在适用的情况下通过流动反应方法来执行。本文所用的术语“分批方法”是指其中将原料在反应器或容器中合并且将产物在反应结束时移出的方法。
[0072]
另外,以下制备中描述的某些中间体可以含有一个或多个氮保护基。可变的保护基在每次出现时可相同或不同,这取决于具体的反应条件和待进行的具体转化。保护和脱保护条件是本领域技术人员公知的,并且描述于文献中(参见例如“greene’s protective groups in organic synthesis”,第四版,peter g.m.wuts和theodora w.greene,john wiley and sons,inc.2007)。
[0073]
当在本文中使用时,以下列出的缩写定义如下:是指埃。“acn”表示乙腈。“acoh”表示乙酸。“bn”表示苄基;“nbuli”表示正丁基锂。“cas no.”是指化学文摘登记号。“dcm”表示二氯甲烷。“dmf”表示n,n-二甲基甲酰胺。“dipea”表示二异丙基乙胺。“dmso”表示二甲亚砜(如果用于nmr,则为全氘化[d6])。“etoac”表示乙酸乙酯。“etoh”表示乙醇。“hbtu”表示(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐。“hplc”表示高效液相色谱。“htrf”表示均相时间分辨荧光。“hr”或“h”表示小时。“ipa”表示异丙醇。“ipc”表示过程质量控制(in-process control)。“lah”表示氢化锂铝。“lcms”表示液相色谱质谱。“lda”表示二异丙基氨基化锂。“me”作为化合物的结构代表中的取代基表示甲基。“meoh”表示甲醇。“min”表示分钟。“ms”表示质谱法或质谱图。“mtbe”表示甲基叔丁基醚。“nmr”表示核磁共振。“nmt”表示不超过。“oac”表示乙酸酯。“psig”表示磅/平方英寸规格。“pybop”表示(六氟磷酸苯并三唑-1-基-氧基三吡咯烷-1-基鏻)。“rt”表示室温/环境温度。“sec”作为时间单位表示秒。“tbs-cl”表示叔丁基二甲基氯硅烷。“tea”表示三乙胺。“thf”表示四氢呋喃。“tr”表示保留时间。“w/w”表示重量与重量的比。
[0074]
下面提供了改进的制备拉司米地坦的路线,为路线i和/或ii,以及下面提供的其它另外的方法。
[0075]“药学上可接受的盐”或“药学上可接受的盐”是指本发明化合物的相对无毒的、无机和有机盐或盐类。本领域技术人员将理解,本发明的化合物能够形成盐。本发明的一些化合物含有碱性杂环,因此与许多无机和有机酸中的任一种反应形成药学上可接受的酸加成盐。所述药学上可接受的酸加成盐和制备它们的常规方法是本领域公知的。参见,例如p.stahl等,handbook of pharmaceutical salts:properties,selection and use,(vcha/wiley-vch,2008);s.m.berge等,“pharmaceutical salts”,journal of pharmaceutical sciences,第66卷,第1期,1977年1月。
[0076]“工艺规模”合成是指制备500mg至1000kg或更多的2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺半琥珀酸盐。优选地,“工艺规模”合成在良好的生产工艺(gmp)或商业生产供人消费的药物产品所需的类似条件下进行。优选地,上述路线i和/或ii的方法中的“工艺规模”是指以至少1千克生产的批次,和/或以至少10千克生产的批次,和/或以至少100千克生产的批次。
[0077]
通用化学
[0078]
流程1
[0079][0080]
流程1描述了一种工艺规模的拉司米地坦半琥珀酸盐化合物i的合成。市售哌啶4-羧酸1的n-甲基化可以在本领域技术人员可确认的各种还原条件下完成,具体地,在过量甲酸中用约1.3当量的甲醛处理仲胺,得到n-甲基哌啶2。二乙酰胺3的形成可以使用常规的酰胺偶联试剂如苯并三唑、hbtu或pybop,或通过将羧酸转化为酰氯,使用本领域公知的试剂如草酰氯或亚硫酰氯来实现。更具体地说,n-甲基哌啶-4-羧酸2可以通过用约1.2当量亚硫
酰氯在约50℃处理1小时,将其转化为酰氯,此时反应混合物可以冷却至约0℃,加入1.5当量二乙胺和3当量三甲胺。将游离碱与hcl一起搅拌,得到二乙酰胺水合盐酸盐3。本领域技术人员可以认识到,吡啶基酮4可以通过用锂化的溴吡啶3a处理二乙酰胺3得到。更具体地,(6-溴-2-吡啶基)锂可通过在约-58℃下用n-buli处理2,6二溴吡啶而形成。单独地,哌啶-4-二乙酰胺盐酸盐水合物3可用约2当量naoh处理,并将所得游离碱在约-58℃下加入锂化物中。所得混合物可用hbr处理,形成吡啶基溴氢溴酸盐4。吡啶基溴氢溴酸盐4的胺化可使用本领域技术人员熟知的过渡金属催化来实现。更具体地,向吡啶溴4中加入约0.075当量的cu2o、约28当量的乙二醇中的nh3,并搅拌至约80℃,将反应物冷却至室温,用h2o猝灭,用20%naoh水溶液洗涤,用20%在ipa中的hcl和少量h2o形成浆液,得到氨基吡啶二水合物二盐酸盐5,为结晶固体。吡啶基苯甲酰胺盐酸盐6可以通过用酰氯5a处理氨基吡啶5的游离碱来制备。更具体地,氨基吡啶二水合物二盐酸盐5可用6%naoh水溶液处理以得到游离碱。另外,2,4,6-三氟苯甲酸可以在约100℃下用亚硫酰氯和上述游离碱5处理,得到吡啶基苯甲酰胺盐酸盐6。半琥珀酸盐i可以通过用约2当量nahco3处理盐酸盐6,然后用约0.55当量琥珀酸处理,得到拉司米地坦半琥珀酸盐化合物i。
[0081]
流程2
[0082][0083]
流程2描述了(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物盐酸盐5的合成。吡啶基溴氢溴酸盐4的胺化可以如流程1所述使用本领域技术人员熟知的过渡金属催化来实现。更具体地说,向吡啶基溴4中加入约0.075当量cu2o、约28当量在乙二醇中的nh3,并在约80℃搅拌,将反应物冷却至室温,用h2o猝灭,用20%naoh水溶液洗涤,用20%在ipa中的hcl和少量h2o形成浆液,得到氨基吡啶二水合物盐酸盐5。
[0084]
流程3
[0085][0086]
流程3说明了合成拉司米地坦半琥珀酸盐i的改进方法。市售哌啶4-羧酸1的n-甲基化可以在本领域技术人员可确认的各种还原条件下完成,具体地,在过量甲酸中用约1.3当量的甲醛处理仲胺,得到n-甲基哌啶2。二乙酰胺3的形成可以使用常规的酰胺偶联试剂如苯并三唑、hbtu或pybop,或通过将羧酸转化为酰氯,使用本领域公知的试剂如草酰氯或亚硫酰氯来实现。更具体地说,n-甲基哌啶-4-羧酸2可以通过用约1.2当量亚硫酰氯在约50℃处理1小时,将其转化为酰氯,此时反应混合物可以冷却至约0℃,加入1.5当量二乙胺和3当量三甲胺。将游离碱与hcl一起搅拌,得到二乙酰胺水合盐酸盐3。本领域技术人员可以认识到,吡啶基酮4可以通过用锂化的溴吡啶3a处理二乙酰胺3得到。更具体地,(6-溴-2-吡啶基)锂可通过在约-58℃下用n-buli处理2,6二溴吡啶而形成。单独地,哌啶-4-二乙酰胺盐酸盐水合物3可用约2当量naoh处理,并将所得游离碱在约-58℃下加入锂化物中。所得混合物可用hbr处理,形成吡啶基溴氢溴酸盐4。可以使用本领域技术人员公知的过渡金属催化来实现吡啶基溴氢溴酸盐4的胺化以获得酰胺6。具体地,如文献中所熟知的,吡啶基酮4可以用合适的无机碱转化为其相应的游离碱形式,并施加buchwald-型偶联条件。更具体地说,化合物4的游离碱可以在合适的非质子溶剂如甲苯或二甲苯中搅拌,其含有约1-5重量%的水、约1.1当量的市售2,4,6-三氟苯甲酰胺(cas#82019-50-9)、约1.5当量的碳酸钾、约0.005至约0.015当量的合适的钯催化剂,如乙酸钯(ii),及约0.01至0.02当量的合适的膦配体化合物如xaphos、xphos或dpephos。所得混合物可以在约70℃下加热约12-24小时。反应混合物可以用水和有机溶剂如dcm或etoac的合适混合物稀释,有机层可以用合适的钯清除剂如硫脲改性的硅胶在约室温至约65℃处理约8-24小时。可以将所得混合物冷却、过
滤、用活性炭处理、过滤并减压浓缩。可以将所得残余物溶于适当的醇溶剂,例如乙醇,并在约55℃用溶解于乙醇中的约0.5当量琥珀酸溶液缓慢处理。所得混合物可以在约10小时内冷却至室温,所得浆液可以通过在一系列加热至60℃和在4小时内冷却回室温的热循环下处理进行浆液研磨。可以通过过滤收集得到的固体,在约40℃干燥约4小时,并任选地进行气流粉碎,获得拉司米地坦半琥珀酸盐i。
[0087]
实验程序
[0088]
以下方法中间体的制备进一步阐述了本发明,并代表了各种化合物的典型合成。试剂和起始材料是容易获得的,或者可以由本领域普通技术人员容易地合成。应当理解,所述制备和实施例是通过举例说明的方式来阐述的,并且本领域普通技术人员可以进行各种修改。
[0089]
lc-es/ms在hp1100液相色谱系统上进行。电喷雾质谱测定(以正和/或负模式获得)在与hp1100 hplc连接的质量选择性检测器四极质谱仪上进行。lc-ms条件(低ph):柱:nx c18 2.1mm
×
50mm,3.0μ;梯度:5-100%b在3分钟内,然后100%b在0.75分钟,柱温:50℃
±
10℃;流量:1.2ml/min;溶剂a:含0.1%hcooh的去离子水;溶剂b:含0.1%甲酸的acn;波长214nm。备选lc-ms条件(高ph):柱:ms c18columns 2.1
×
50mm,3.5μm;梯度:5%溶剂a进行0.25分钟,3分钟内5%-100%溶剂b的梯度,100%溶剂b进行0.5分钟,或3分钟内10%-100%溶剂b和100%溶剂b进行0.75分钟;柱温:50℃
±
10℃;流量:1.2ml/min;溶剂a:10mm nh4hco
3 ph 9;溶剂b:acn;波长:214nm。
[0090]
nmr谱在bruker aviii hd 400或500mhz nmr波谱仪上进行,作为cdcl3或(cd3)2so溶液获得,以ppm为单位报道,使用残余溶剂[cdcl3,7.26ppm;(cd3)2so,2.05ppm]作为参考标准。当报告峰多重性时,可以使用以下缩写:s(单峰)、d(双峰)、t(三重峰)、q(四重峰)、m(多重峰)、br-s(宽单峰)、dd(双重峰)、dt(双重峰或三重峰)。当报道时,偶合常数(j)以赫兹(hz)报道。
[0091]
氯化物分析在esaplus仪器上进行,所述仪器配备有(带电气溶胶检测器)-hplc,acclaimtrinity p1(100x 3.0mm,3um),流动相:50mm乙酸铵,acn中ph~5。
[0092]
本文所述的化合物可通过本领域技术人员已知的一般方法或通过本文所述的方法制备。这些流程的步骤的合适反应条件是本领域公知的,并且溶剂和共试剂的适当取代是本领域技术人员已知的。同样,本领域技术人员应理解,合成中间体可根据需要或期望通过各种公知技术分离和/或纯化,并且通常,可在随后的合成步骤中直接使用各种中间体而几乎不纯化或不纯化。此外,技术人员将理解,在一些情况下,引入结构部分的顺序不是关键的。
[0093]
制备1
[0094]
1-甲基哌啶-4-甲酸盐酸盐
[0095][0096]
流程1,步骤a:向带夹套的反应器中加入哌啶-4-甲酸(10.0g,77.5mmol)和去离子水(40ml)。将该混合液加热至回流(95
–
100℃)。经30分钟加入甲酸(13.9g,302mmol)。经至少30分钟向混合物中滴加37%甲醛水溶液(8.1g,101mmol)。将水(0.3ml)用作进入反应器的管线冲洗液。将该混合液在回流下(95
–
100℃)搅拌4小时,并通过hplc取样进行ipc分析(nmt 0.5%哌啶-4-甲酸)。如果哌啶-4-羧酸的量高于0.5%,则将混合物再搅拌2小时。如果符合规格,将溶液在真空下浓缩,直到剩余~20ml的残余体积,并将残余物冷却至45-50℃,经不少于30分钟向冷却的溶液中加入33%hcl水溶液(12.8g,116mmol)。将水(0.3ml)用作进入反应器的管线冲洗液。真空蒸馏除去水,直到剩余~20ml的残余体积。在45
–
50℃向浓缩液中加入acn(42.4ml),并将混合物在大气压下浓缩至剩余~40ml的残余体积。向浓缩液中在45
–
50℃加入acn(20.4ml),并将该混合液在大气压下浓缩至残余体积为~40ml。重复稀释/浓缩操作,直至karl-fischer分析对水分含量的过程控制为nmt 0.2%;在这些操作期间形成浆液。在45-50℃向该浆液中加入acn(10.2ml),经1小时将该浆液冷却至20℃并再搅拌2小时,通过过滤分离所得固体,用acn(10.2ml)冲洗滤饼。将湿滤饼在40℃在氮气和大气压下干燥,得到标题化合物(12.1g,87%收率)。ms(m/z):144(m h)。
[0097]
制备2
[0098]
n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合物盐酸盐
[0099][0100]
流程1,步骤b:向带夹套的反应器中加入1-甲基哌啶-4-甲酸盐酸盐(30.0g,167mmol)、氯苯(240ml)和dmf(0.61g,8.35mmol),并将得到的混合液加热至50℃。经1小时时间向该热混悬液中加入亚硫酰氯(24.2g,200.4mmol)。将氯苯(13.5ml)用作进入反应器的管线冲洗液。亚硫酰氯添加完成后将该混合液搅拌5小时。然后将该溶液冷却至0至10℃。经3小时将由二乙胺(17.7g,12.5mmol)和tea(50.7g,25mmol)制备的溶液加入冷的反应混合物中。将氯苯(13.5ml)用作进入反应器的管线冲洗液。在胺混合物完全加入后,将混合物搅拌2小时。将反应物用20重量%naoh水溶液(180.3g,902mmol)处理,并在室温搅拌2小时。将水(3ml)用作进入反应器的管线冲洗液。使混合物沉降2小时,除去水相。将剩余的有机相置于真空下。加热混合物以蒸除残余胺以及大部分氯苯。在收集了大约十体积的馏出物后,
使用氮气将反应器排空至大气压。将剩余溶液冷却至10℃至30℃之间,将thf(120ml)和水(4.54g,252mmol)加入反应器中。在室温,通过加入20重量%hcl的异丙醇溶液(30.4g,167mmol)使反应混合物沉淀出所需产物。将thf(5.4ml)用作进入反应器的管线冲洗液。在hcl完全加入后,将混悬液在室温搅拌2小时。过滤收集所得固体,并用thf(75.0ml洗涤)。将收集的固体在40℃真空干燥16小时,得到标题化合物(35.5g,84%收率)。ms(m/z):199(m h)。
[0101]
制备3
[0102]
(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮氢溴酸盐
[0103][0104]
流程1,步骤c:用20重量%的naoh(34.0g,170mmol)水溶液处理n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合物盐酸盐(21.5g,85.1mmol)在mtbe(109ml)中的混悬液。用水冲洗(1.94ml)以完成添加。将混合物在室温搅拌30分钟,使各相沉降,并分离各相。将水相用mtbe萃取(43.7ml),合并有机相。在大气压下通过蒸馏干燥有机相,直到通过karl-fischer分析对水含量的过程控制小于0.10重量%。如果不满足目标分析,向反应物中加入mtbe(43.7ml),并重复蒸馏。通常需要三次蒸馏以达到对水的目标分析。在单独的反应器中,加入2,6-二溴吡啶(30.2g,128mmol)和mtbe(105ml)的混合液,并冷却至低于
–
58℃。经2小时向冷却的混悬液中加入2.5m的n-buli的己烷溶液(51.3ml,128mmol)。用mtbe(4.5ml)的冲洗液完成转移。在完成n-buli添加后,将混合物老化,同时将温度保持在低于-58℃下另外2小时。老化后,经45分钟时间将n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合物盐酸盐在mtbe中的溶液加入冷反应物中。用mtbe(13.5ml)的冲洗液完成该转移。在完全加入在mtbe中的n,n-二乙基-1-甲基-哌啶-4-甲酰胺水合物盐酸盐后,将该混合液老化至少30分钟。老化后,经1小时将反应温至0℃。以一定速率将该冷的反应混合液加入2.5m hcl水溶液(146ml,366mmol)中以保持淬灭温度在nmt 30℃。用mtbe(13.5ml)的冲洗液完成转移。在转移完成后,将该混合液搅拌至少30min,并使各相沉降。分离各相,并保留水相。将n-buoh(54.8ml)加入水相中,并将该混合液用20重量%的naoh(59.5g,298mmol)水溶液处理。用水(2.80ml)冲洗以完成转移。将该混合液搅拌至少30分钟,并使各相沉降。分离各相,并保留有机相。将水相用n-buoh萃取(54.8ml)。将合并的有机相通过真空蒸馏干燥,以获得通过karl-fischer分析<0.20重量%的水含量的过程控制。如果不满足目标分析,则加入n-buoh(41.1ml),并重复蒸馏。通常,需要两次蒸馏以达到过程控制目标分析。通过过滤澄清浓缩的溶液,用n-buoh(89.6ml)冲洗以完成转移,并冲洗过滤器。用48重量%的hbr水溶液(9.91ml,87.7mmol)处理澄清的溶液90分钟。用正丁醇(13.8ml)冲洗以完成转移。ph的检测显示反应混合物的ph为~1。在大气压下通过蒸馏干燥该混合物,以获得通过karl-fischer
分析<0.30重量%的水含量的过程控制。将混合物浓缩至172ml。如果不满足目标分析,则加入n-buoh(54.8ml),并重复蒸馏。将混合物冷却至20℃并搅拌12小时。通过过滤收集所得固体,并用n-buoh(10.75ml)洗涤两次。将固体在60℃真空干燥,得到标题化合物(24.8g,80%收率)。ms(m/z):283,285(
79
br,
81
br,m h)。
[0105]
制备4
[0106]
(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐
[0107][0108]
流程1,步骤d:向压力反应器中加入(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮氢溴酸盐(30g,82.9mmol)和cu2o(880mg,6.2mmol)。用氮气/真空吹扫循环交换顶部空间三次。向固体中加入nh3/乙二醇溶液(总共273.5g,39.1g nh3,2.33mol;210ml乙二醇),并将得到的混合液在室温搅拌2小时。将该混合液加热至80℃,搅拌10小时,冷却至室温,对(6-溴-2-吡啶基)-(1-甲基-4-哌啶基)甲酮氢溴酸盐nmt 2%进行过程控制取样。如果不满足目标分析,则将反应物在80℃下再搅拌4小时并再次取样。向完成的反应中加入h2o(90ml),并将该混合液过滤。将滤液加入nacl水溶液(253.9g nacl,2.73mol,13.7l/kg h2o)中,并将得到的混合液在室温搅拌10分钟。向该混合物中加入20%naoh水溶液(4.44当量,368mmol),并将该两相混合物在室温搅拌5分钟。将该混合液在室温用mtbe萃取(90ml)4
–
5次。用5重量%的碳处理合并的mtbe层30分钟,并通过过滤除去碳。在真空下将有机滤液浓缩至~150ml。向浓缩的滤液中加入ipa(200ml),并将溶液在真空下浓缩至~150ml两次。根据需要重复ipa蒸馏以满足用于水的过程控制的目标分析。通过karl-fischer分析确定水含量不超过0.2%。在室温,在另一个反应器中加20重量%hcl在ipa中的溶液(30g,166mmol)和水(10.5ml)室温。将浓缩的产物混合物在90分钟内加入hcl溶液中。将所得浆液在室温下搅拌不少于8小时。过滤浆液,用95:5ipa/h2o(36ml)的混合物在室温洗涤两次,在40℃下真空干燥16
–
24小时,得到标题化合物(18.4g 68%收率)。ms(m/z):220(m h)。1h nmr(400mhz,d2o/dmso-d6)δppm 1.74-1.88(m,2h),2.05(br d,j=14.9hz,2h),2.73(s,3h),3.01(td,j=13.1,2.6hz,2h),3.41-3.50(m,2h),3.55(tt,j=12.0,3.5hz,1h),7.14(dd,j=9.0,0.7hz,1h),7.59(d,j=7.2hz,1h),7.90(dd,j=9.0,7.2hz,1h)。
13
c nmr(101mhz,d2o//dmso-d6)δppm 27.4,40.7,44.6,54.6,117.2,121.1,137.9,145.2,156.2,196.3。氯化物分析:20.23%(n=2)。
[0109]
x射线粉末衍射(xrpd)的结晶形式
[0110]
在bruker d4 dearter x-射线粉末衍射仪上获得结晶固体的xrpd图,该衍射仪配备有cukα源和vantec检测器,在35kv和50ma下操作。在4和40 2θ
°
之间扫描样品,步长为4和40 2θ
°
,扫描速率为0.5秒/步,使用1.0mm的发散度、6.6mm的固定抗散射和11.3mm的检测器
狭缝。将干燥粉末装在石英样品架上,并使用载玻片获得光滑表面。在环境温度和相对湿度下收集晶形衍射图。在mdi-jade中,根据内部nist 675标准物,在完整图案移动后,确定晶体峰位置,峰值在8.853和26.774 2θ
°
。在晶体学领域中众所周知,对于任何给定的晶型,衍射峰的相对强度可能由于由诸如晶体形态和习性等因素导致的优选取向而变化。当存在优选取向的影响时,峰强度改变,但是多晶型物的特征峰位置不变。参见例如美国药典#23,国家处方集#18,第1843页-1844页,1995。此外,在结晶学领域中也众所周知,对于任何给定的晶型,角峰位置可以稍微变化。例如,峰位置可能由于分析样品的温度变化、样品位移或存在或不存在内标而偏移。在本情况中,
±
0.2 2θ
°
的峰位置变化被认为考虑了这些潜在变化,而不妨碍所指示的晶型的明确鉴定。可以基于区别峰的任何独特组合来确认晶型。
[0111]
制备4的样品,(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐,其特征在于使用cukα辐照的xrd图具有下表1中所述的衍射峰(2θ值),尤其是具有在8.3
°
处的峰以及选自16.6
°
、23.5
°
和33.7
°
的一个或多个峰,衍射角的容许误差为0.2
°
。
[0112]
表1:制备4的结晶化合物的x-射线粉末衍射峰;(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二水合物二盐酸盐
[0113][0114][0115]
制备5
[0116]
2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺盐酸盐
[0117][0118]
流程1,步骤e:向(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二盐酸盐二水合物(10g,30.6mmol)在氯苯(65ml)中的混悬液中加入6w/w%naoh水溶液(3g,75mmol)。将该两相混合物在搅拌下加热至54℃达30分钟,使该混合液在30分钟内分离,并在54℃下分离各
层。将水层用氯苯(45ml)在室温反萃取。合并有机层,并在真空下蒸馏至~62ml,得到(6-氨基吡啶-2-基)(1-甲基哌啶-4-基)甲酮溶液。在单独的反应器中加入2,4,6-三氟苯甲酸(5.9g,1.1当量,33.7mmol)、dmf(62mg,0.85mmol)和氯苯(32ml),并将混合物加热至80℃。在80℃下经4小时向加热的混合物中加入亚硫酰氯(4.37g,37mmol),将混合物在80℃下搅拌至少6小时,并加热至100℃达至少6小时以清除残余的hcl气体。将酰基氯溶液冷却至室温,并转移到单独的反应器中。将酰基氯溶液加热至100℃,并经4小时加入(6-氨基吡啶-2-基)(1-甲基哌啶-4-基)甲酮。将所得浆液在100℃下再搅拌3小时,并冷却至室温。向冷却的浆液中加入acn(100ml)。将所得浆液加热至80℃达1小时,并在2小时内冷却至室温。将所得浆液在室温下再搅拌1小时,并过滤。在室温用acn(10ml)洗涤滤饼。将收集的固体在真空下在100℃干燥16小时,得到标题化合物(10.7g,85%收率)。ms m/z 378(m h)。
[0119]
制备5的备选方法
[0120]
向(6-氨基-2-吡啶基)-(1-甲基-4-哌啶基)甲酮二盐酸盐二水合物(10g,30.6mmol)在氯苯(65ml)中的混悬液中加入6w/w%naoh水溶液(2.97g,74.4mmol)。将两相混合物在搅拌下加热至54℃达30分钟,并使各层在30分钟内分离。在54℃分离各层,将水层在室温下用氯苯(45ml)反萃取。合并有机层,并在真空下蒸馏至~62ml,得到(6-氨基吡啶-2-基)(1-甲基哌啶-4-基)甲酮溶液。在另一反应器中,加入2,4,6-三氟苯甲酸(5.9g,33.7mmol)、dmf(62mg,0.85mmol)和氯苯(32ml),将混合物加热至80℃。在80℃经4小时向加热的混合物中加入亚硫酰氯(4.4g,37mmol)。将混合物在80℃搅拌至少6小时,并加热至100℃至少6小时以清除残余的hcl气体。将酰基氯溶液冷却至室温,并转移到单独的反应器中。将酰基氯溶液加热至100℃,经4小时向溶液中加入(6-氨基吡啶-2-基)(1-甲基哌啶-4-基)甲酮。将所得浆液在100℃再搅拌3小时,并冷却至室温。向冷却的浆液中加入acn(100ml)。将所得浆液加热至80℃达1小时,并在2小时内冷却至室温。将所得浆液在室温下再搅拌1小时,过滤收集所得固体。在室温下用acn(10ml)洗涤滤饼。将固体在真空下在100℃干燥16小时,得到标题化合物(10.7g,85%收率)。ms m/z 378(m h)。
[0121]
制备6
[0122]
2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺半琥珀酸盐
[0123][0124]
流程1,步骤f:向反应器中加入2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺盐酸盐(20g,48.4mmol)和mtbe(202ml)。在室温下经1小时向搅拌的浆液中加入nahco3水溶液(8.13g,96.8mmol nahco3在200ml水中)。分离两相混合物,将水层用mtbe(202ml)反萃取。将合并的有机层在真空下蒸馏至终体积为~200ml。向蒸馏的溶液中加入sio2(2g),将所得混合物在室温搅拌30分钟,过滤,将滤饼用mtbe(10.8ml)冲洗。向滤液中加入碳(340mg;或者,溶液可以通过碳柱过滤),将所得混合物在室温搅拌30分钟,通过1-5μ
三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺溶液中,并将所得溶液在55℃搅拌,将2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺半琥珀酸盐作为晶种以固体或在甲苯变性etoh中的浆液形式加入。经1.5小时将剩余的在甲苯变性etoh溶液中的琥珀酸转移至反应器中。将反应器的内容物经10小时冷却至室温。可以将所得浆液研磨以控制粒度。如果研磨浆液,反应器的内容物可经历一系列热循环,其通过在搅拌下加热至60℃并在4小时内冷却回室温进行,以进一步控制粒度分布。过滤浆液,用etoh(100ml,用甲苯变性)冲洗,并在40℃减压干燥12小时,得到标题化合物(43.9g,73%收率)。然后可以将干燥的固体喷射研磨以进一步控制粒度。
[0129]
药品制备方法的描述
[0130]
在一个实施方案中,通过本文提供的方法制备的拉司米地坦可进一步制备为某些有用的药物产品形式。在一个实施方案中,所述药物产品形式可以是椭圆形的50和100mg的凹陷的、水性薄膜包衣的即释剂。50mg片剂是浅灰色的椭圆形片剂,一侧凹陷图案为“4312”,另一侧凹陷图案为“l-50”。100mg片剂是浅紫色、椭圆形的片剂,一侧凹陷图案为“4491”,另一侧凹陷图案为“l-100”。
[0131]
下列单元配方可用于生产拉司米地坦片剂。成分命名惯例基于usp。
[0132]
表2。拉司米地坦50mg和100mg片剂的单元配方
[0133][0134]
表2注释:
[0135]
a.使用0.86469的盐转化因子计算拉司米地坦半琥珀酸盐的量。可以相应地调节微晶纤维素的量以保持目标片剂重量。
[0136]
b.在制粒操作中使用净化水。随后在干燥操作期间除去大部分水。
[0137]
c.干燥过程后残留了少量的残余水,其可以是游离水或与药物物质结合的水合水的形式。
[0138]
d.纯化水用于包衣单元操作。包衣混悬液由20%w/w固体组成。喷涂足够的包衣以达到3%的重量增加。在包衣单元操作期间去除这些水。
[0139]
片剂制备:
[0140]
使用如下所述的高剪切湿法制粒方法制备拉司米地坦片剂。高剪切湿法制粒:将十二烷基硫酸钠通过安全筛,并加入到纯化水中以形成制粒液体。将拉司米地坦药物物质和要湿法制粒的赋形剂(微晶纤维素、预胶化淀粉、交联羧甲基纤维素钠)通过安全筛并在制粒机中混合。在添加制粒液体之前,用制粒机的主叶轮混合材料。在粉末混合的同时,通过加入制粒液体,在制粒机中将粉末共混物制粒。在完成液体添加时,将颗粒湿法成团以促
n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺的共振积分,分别得到29094和9508的面积。乙酸盐与2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺的摩尔比通过取这些面积的比率计算,说明共振质子计数的差异,得到观察到的摩尔比29094/(3x 9508)=1.02乙酸盐:2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺。参见例如图1,其显示了含有马来酸(内标)的拉司米地坦乙酸盐的1h nmr谱(400mhz,dmso-d6)。该结果提供了实验证据,即2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺乙酸盐的制备实施例是单乙酸盐。
[0151]
2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺乙酸盐的x-射线粉末衍射(xrpd)
[0152]
在bruker d4 dearter x-射线粉末衍射仪上获得2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺乙酸盐结晶固体的xrpd图,该衍射仪配备有cukα源和vantec检测器,在35kv和50ma下操作。在4和40 2θ
°
之间扫描样品,步长为4和40 2θ
°
,扫描速率为0.5秒/步,使用1.0mm的发散度、6.6mm的固定抗散射和11.3mm的检测器狭缝。将干燥粉末装在石英样品架上,并使用载玻片获得光滑表面。在环境温度和相对湿度下收集晶形衍射图。在mdi-jade中,根据内部nist 675标准物,在完整图案移动后,确定晶体峰位置,峰值在8.853和26.774 2θ
°
。在晶体学领域中众所周知,对于任何给定的晶型,衍射峰的相对强度可能由于由诸如晶体形态和习性等因素导致的优选取向而变化。当存在优选取向的影响时,峰强度改变,但是多晶型物的特征峰位置不变。参见例如美国药典#23,国家处方集#18,第1843页-1844页,1995。此外,在结晶学领域中也众所周知,对于任何给定的晶形,角峰位置可以稍微变化。例如,峰位置可能由于分析样品的温度变化、样品位移或存在或不存在内标而偏移。在本情况中,
±
0.2 2θ
°
的峰位置变化被认为考虑了这些潜在变化,而不妨碍所指示的晶型的明确鉴定。可以基于区别峰的任何独特组合来确认晶型。
[0153]
制备的结晶乙酸盐的样品的特征在于使用cuka辐照的xrpd图,其具有如下表3中所述的衍射峰(2θ值),尤其是具有在26.2处峰以及选自20.4、14.0和17.9的一个或多个峰;衍射角的容许误差为0.2度。
[0154]
表3。结晶2,4,6-三氟-n-[6-(1-甲基哌啶-4-羰基)-2-吡啶基]苯甲酰胺乙酸盐的x-射线粉末衍射峰
[0155][0156]
拉司米地坦乙酸盐的溶解度及其在皮下注射制剂中的应用:
[0157]
对于皮下制剂的制备,发现拉司米地坦乙酸盐令人惊讶地优于许多替代的盐形式,其在接近生理的液体中以最小体积递送拉司米地坦的剂量。发现拉司米地坦乙酸盐能够在小于或等于约1ml的最小体积中达到约50mg的所需剂量目标,同时达到接近中性的所需目标ph,并且还相对等渗且物理和化学稳定。使用拉司米地坦半琥珀酸盐,实验上确定在接近中性ph时,不使用共溶剂难以达到》50mg/ml的溶解度。为了测定溶解度,用相应的酸和盐制备10mmol缓冲液,并通过改变酸/盐比例来调节ph。过量的固体在室温平衡过夜,通过hplc分析溶液浓度,通过xrpd表征固体。相反,对于拉司米地坦乙酸盐,发现在接近中性ph时,无需调节ph即可达到》50mg/ml的溶解度。
[0158]
表4:
[0159]
拉司米地坦乙酸盐在乙酸盐、柠檬酸盐或磷酸盐缓冲液中的溶解度。用相应的酸和盐制备10mmol的缓冲液,通过改变酸/盐比例调节ph。所有加入的溶质进入溶液中形成粘稠溶液。
[0160]
[0161][0162]
已发现拉司米地坦乙酸盐惊人地显示出高度有利的各药物特性的组合。对于所需的单位剂量,拉司米地坦乙酸盐能够提供所需的溶解度以提供具有小于或等于约1ml剂量体积的高浓度制剂,这对于临床应用如在可获得的自动注射器装置中的使用是很关键的。此外,拉司米地坦乙酸盐以50mg/ml的溶解导致接近中性的ph(ph大约6.8),其是等渗的,并且保持稳定至少2个月。拉司米地坦乙酸盐显示了比具有符合需要的ph特性的拉司米地坦半琥珀酸盐显著更高的溶解度,使得能够以约1ml或更少的体积递送所需的单位剂量。
[0163]
这些结果表明,拉司米地坦乙酸盐能使拉司米地坦的含水溶液具有惊人的高浓度,具有对用于临床肠胃外施用、例如皮下注射而言的有用的药学性质。拉司米地坦的药理学活性是充分确定的(curto,m等人,profiling lasmiditan as a treatment option for migraine,expert opinion on pharmacotherapy(2020),21卷,2期,147-153页)。优选地,皮下注射通过预填充注射器或自动注射器施用,使用技术人员已知的装置(参见例如stauffer vl等,comparison between prefilled syringe and autoinjector devices on patient-reported experiences and pharmacokinetics in galcanezumab studies.,patient prefer adherence.(2018)12:1785-1795和van den bemt bjf等人,a portfolio of biologic self-injection devices in rheumatology:how patient involvement in device design can improve treatment experience.,drug deliv.(2019),26(1):384-392)。这些形式提供固定剂量,而不需要患者测量,提供了剂量准确性和安全性,同时使得患者能够自主操作。使用拉司米地坦乙酸盐以诸如自动注射器的形式用于急性治疗偏头痛发作,为公共机构患者提供了改进的工具,例如在医院的紧急情况下,其中患者使用片剂受到偏头痛发作和相关的恶心和呕吐的破坏,并且患者和/或提供者优选使用改进的可注射形式的拉司米地坦。预期拉司米地坦乙酸盐肠胃外制剂提供直接释放,能够使得起效时间迅速,并且当在偏头痛发作开始时根据需要使用时,相对于口服剂型可以优选允许更短的起效时间。
[0164]
提供用于在中性和生理ph(约6.0-7.5)注射的且与生理液体等渗的(例如280-300mosm/kg)拉司米地坦制剂在临床上是高度理想的,并被认为使例如注射时疼痛和/或组织刺激的可能性最小化。实现约1ml或更少的注射体积使得能够使用诸如自动注射器的可用注射器技术,并且例如在注射时间和/或注射时的疼痛方面为患者提供改善的注射和递送体验。能够使用预填充注射器、笔和/或自动注射器技术对于偏头痛患者在临床上是重要的,因为这些装置在偏头痛发作期间提供了使用便利性,此时患者在产品使用时经常处于束缚下(duress)。此外,能够使用预填充的注射器、笔和/或自动注射器技术在临床上是重要的,因为它们提供了在任何时间便携的药物,这在日常生活过程中是可以轻便获得的(偏头痛发作可以在任何时间发生)。
[0165]
下面的单元配方可以用于生产注射用拉司米地坦溶液。
[0166]
表5。用于自动注射器中的拉司米地坦50mg溶液的单元配方
[0167]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。